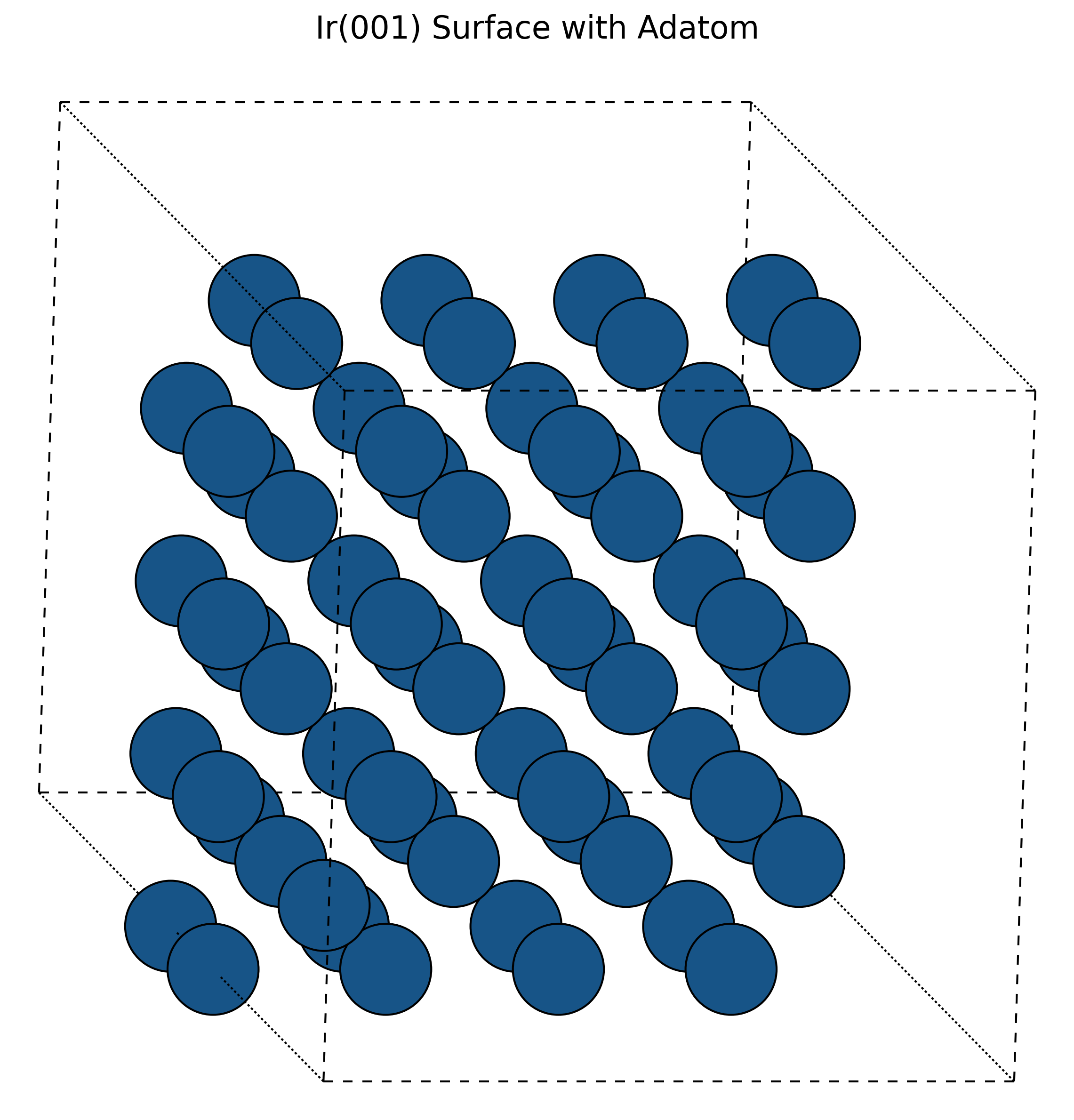
MD Simulation of Self-Diffusion on Metal Surfaces (1994)
A molecular dynamics investigation using EAM and many-body potentials to elucidate atomic exchange mechanisms on Iridium surfaces, verifying Field Ion Microscope observations.
A molecular dynamics investigation using EAM and many-body potentials to elucidate atomic exchange mechanisms on Iridium surfaces, verifying Field Ion Microscope observations.
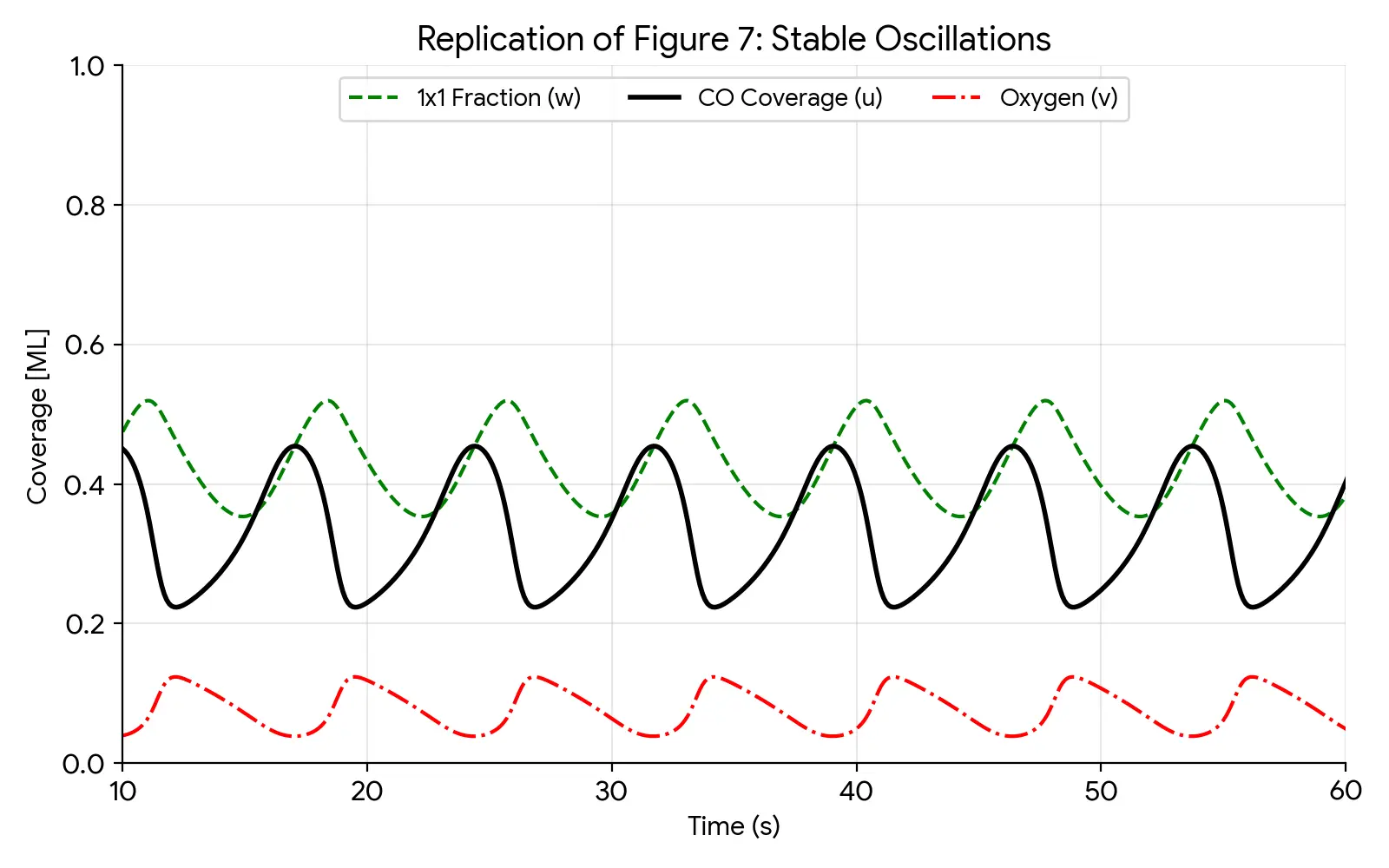
This paper presents a 4-variable kinetic model coupling surface reaction dynamics with structural phase transitions to reproduce complex oscillatory behavior on Pt(110).
Proposes the Hyperbolic Algorithm for Euclidean field theory simulations. By adding a second-order fictitious time derivative to the Langevin equation, the method reduces systematic errors from O(ε) down to O(ε²).
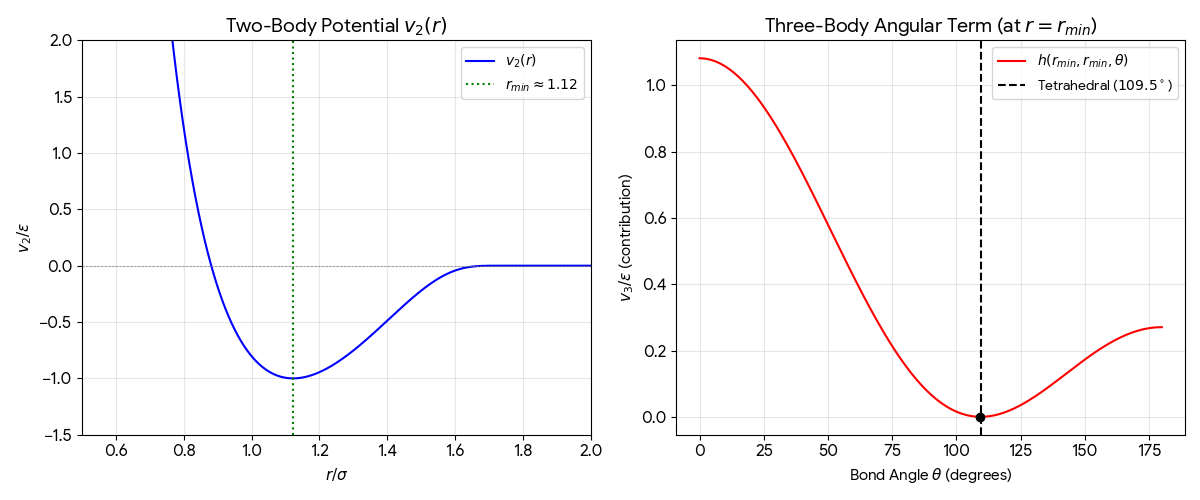
Stillinger and Weber propose a 3-body interaction potential that stabilizes the diamond crystal structure of silicon and reproduces liquid properties through molecular dynamics, addressing the inability of standard pair potentials to model tetrahedral semiconductors.
This molecular dynamics study reveals that adatom dimers on fcc(111) surfaces exhibit simultaneous multiple jumps at intermediate temperatures, migrating with mobility comparable to single adatoms.
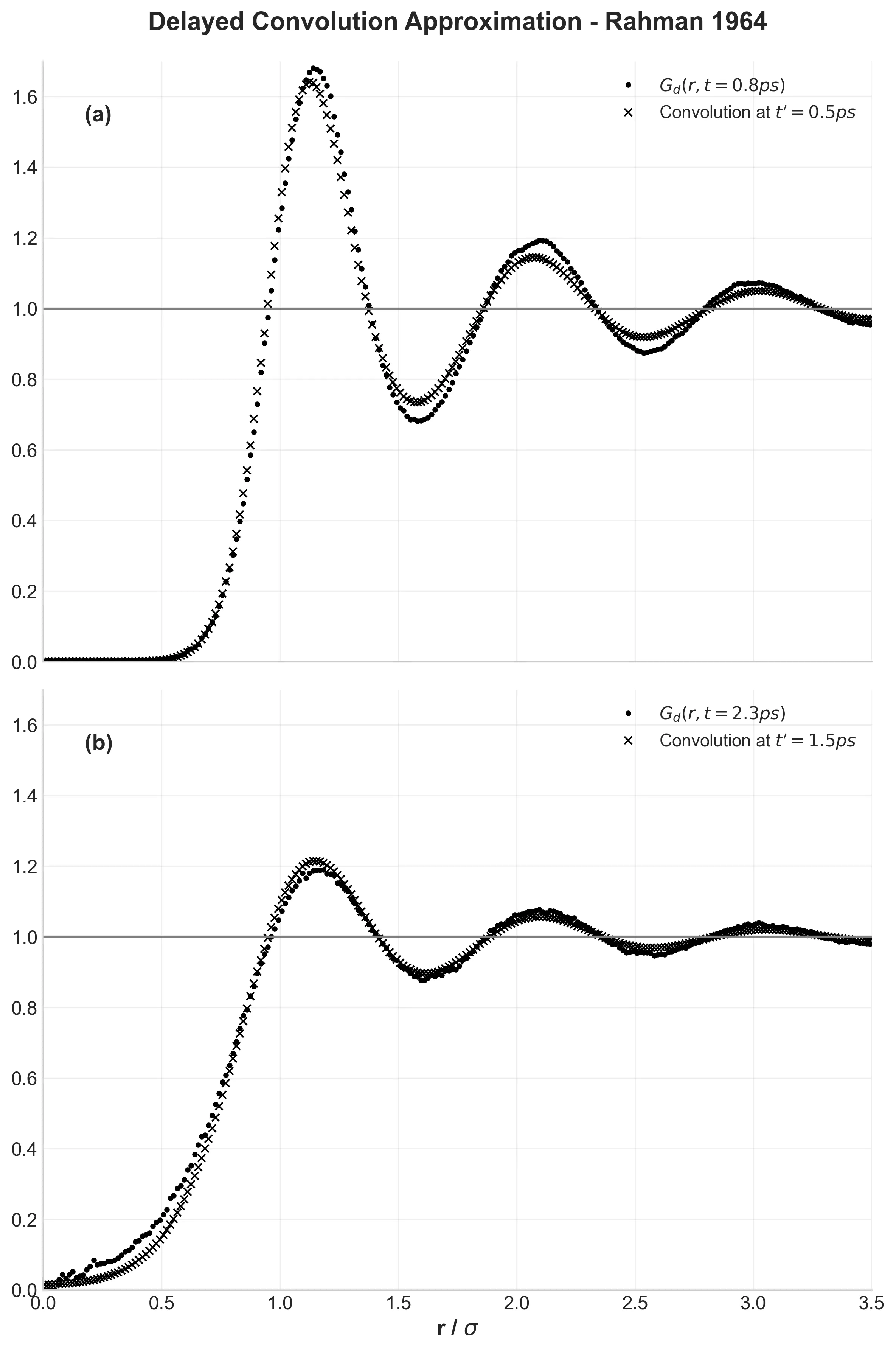
This work validated classical Molecular Dynamics for simulating liquids, revealing the ‘cage effect’ in velocity autocorrelation and establishing predictor-corrector integration algorithms for N-body problems.
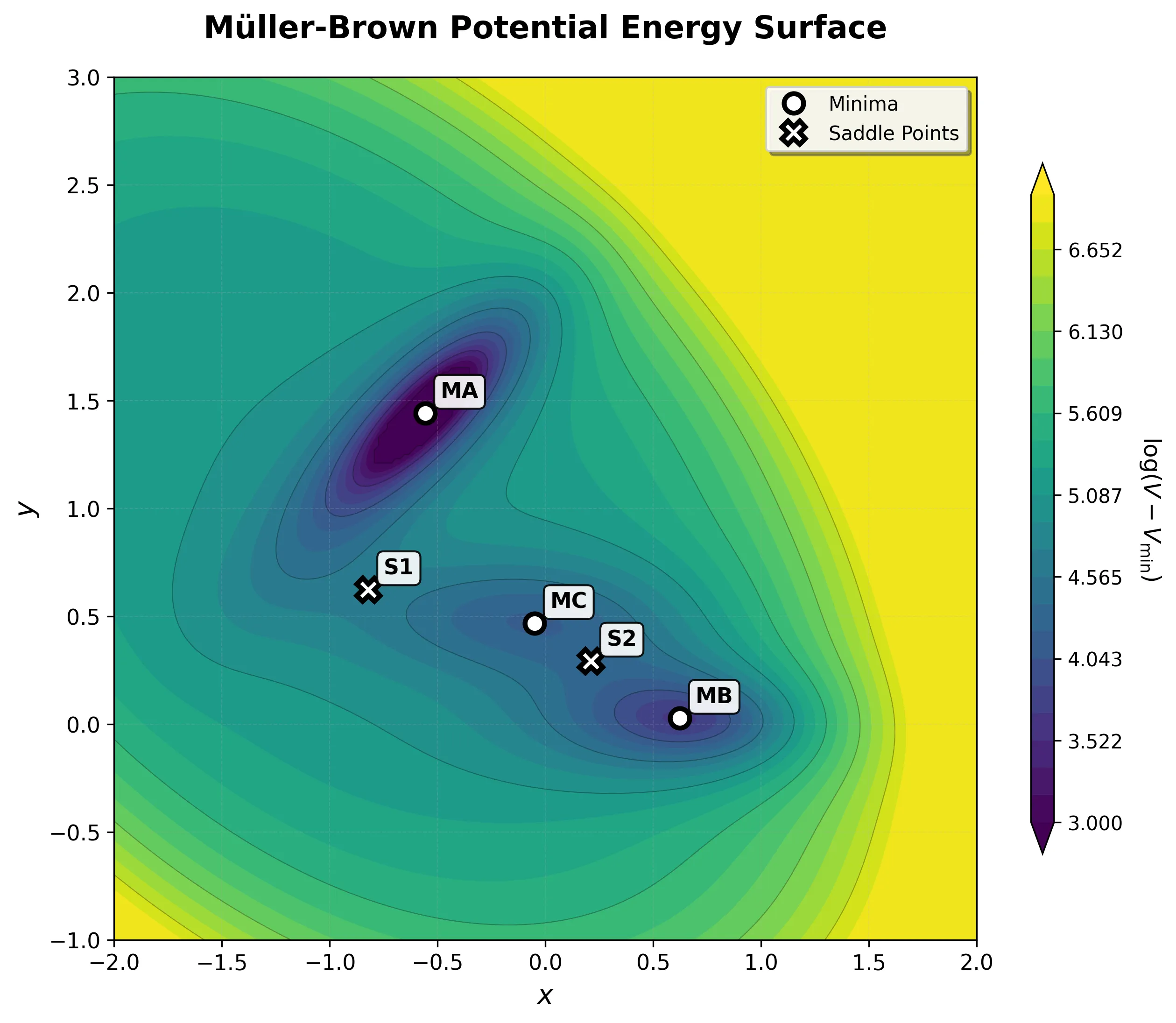
A two-dimensional analytical potential energy surface introduced in 1979 for testing optimization algorithms. It features three minima and curved transition pathways that evaluate an algorithm’s ability to navigate non-trivial topologies.
Step-by-step implementation of the classic Müller-Brown potential in PyTorch, with performance comparisons between analytical and automatic differentiation approaches for molecular dynamics and machine learning applications.
Observe confined particle motion in the deep reactant well of the Müller-Brown potential. This simulation demonstrates thermal motion within a stable energy minimum at -146.70 kJ/mol.
Watch particle dynamics in the product minimum of the Müller-Brown potential. This simulation shows intermediate thermal motion behavior at -108.17 kJ/mol energy level.
A high-performance, GPU-accelerated PyTorch testbed for ML-MD algorithms featuring JIT-compiled analytical Jacobian force kernels achieving 3-10x speedup over autograd, robust Langevin dynamics with Velocity-Verlet integration, and modular architecture designed as ground-truth validation for novel machine learning approaches in molecular dynamics.
Experience rare transition events between energy basins in this extended Müller-Brown simulation. Watch as particles overcome energy barriers to explore different regions of the potential energy landscape.