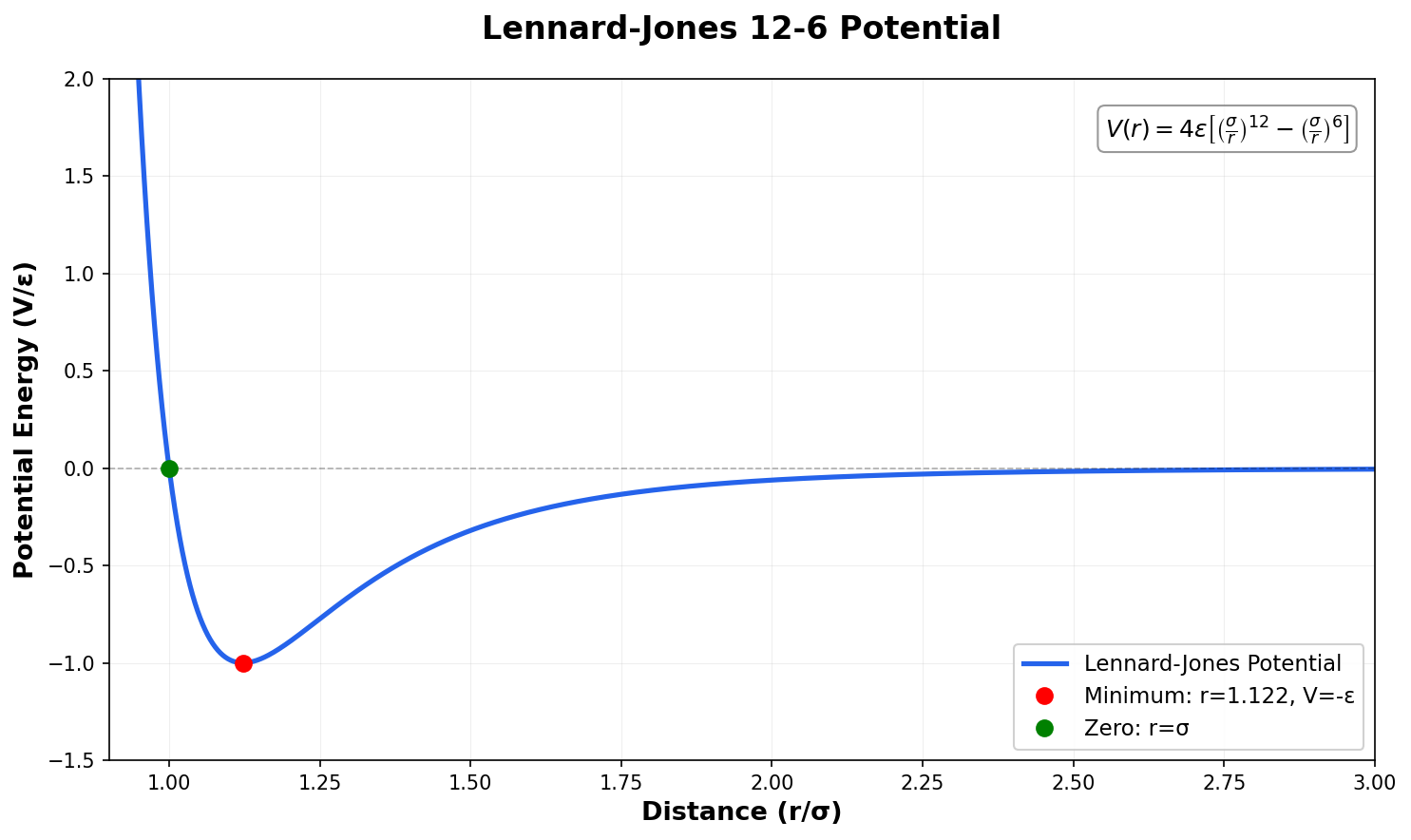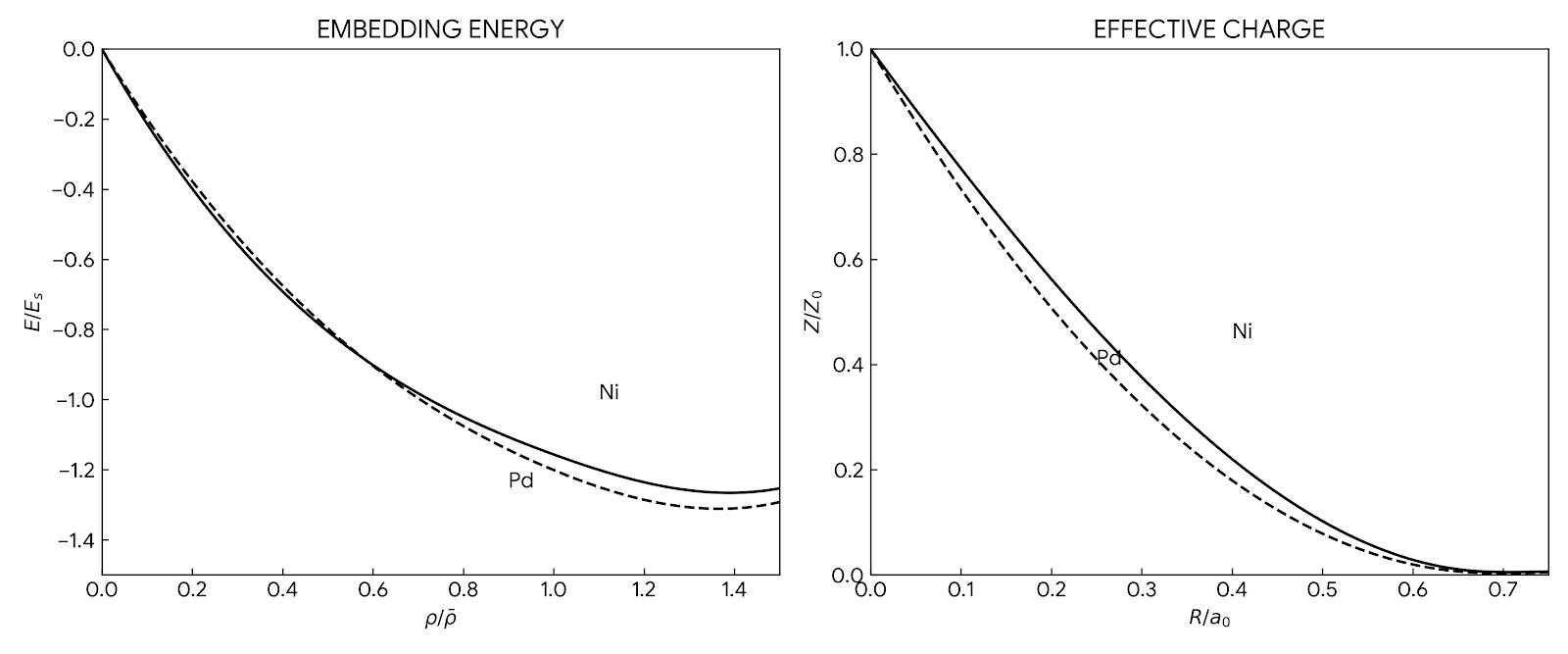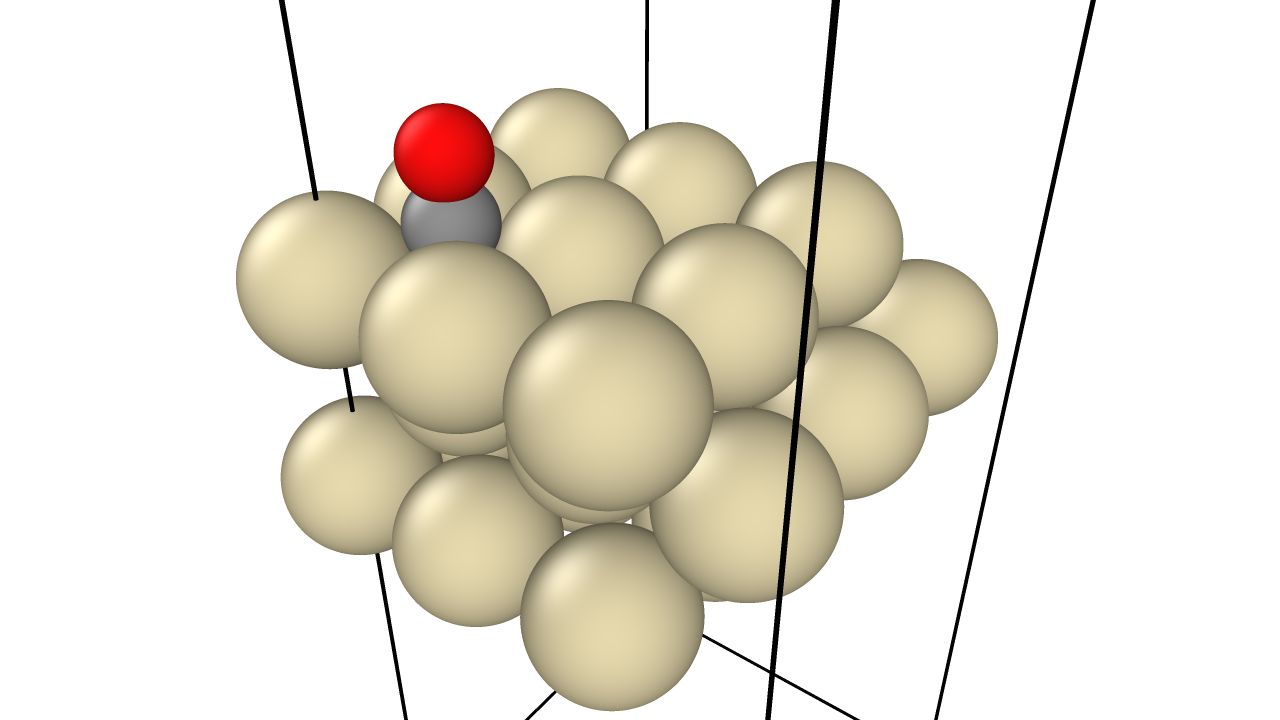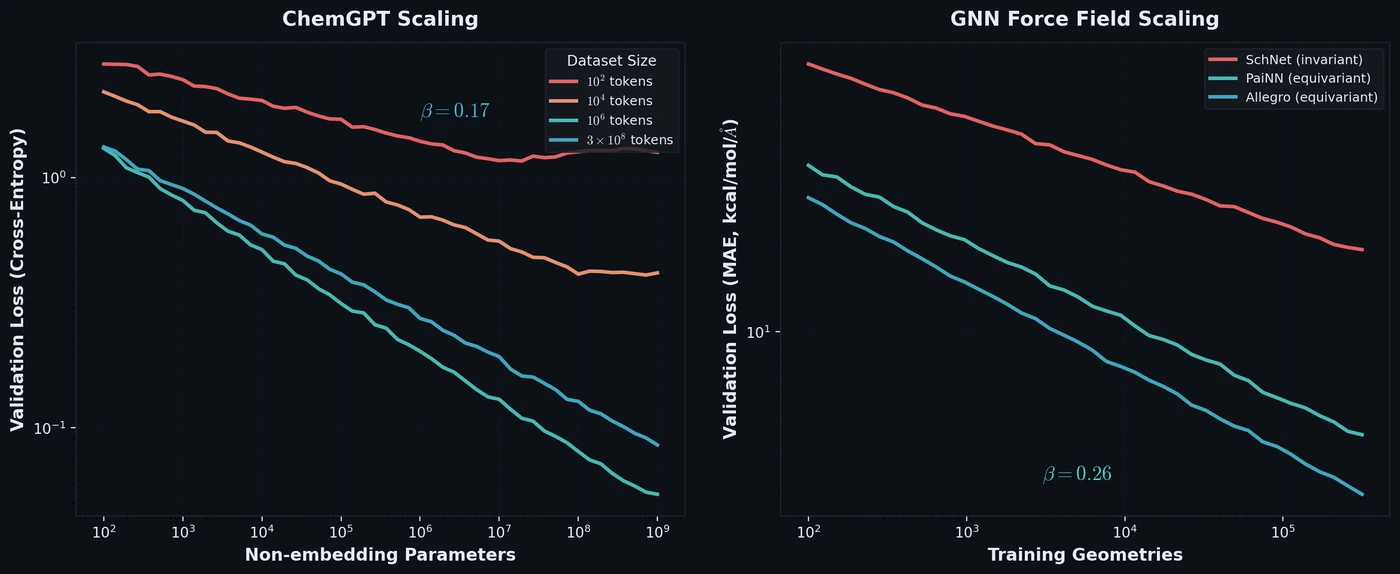
Neural Scaling of Deep Chemical Models
Frey et al. discover empirical power-law scaling relations for both chemical language models (ChemGPT, up to 1B parameters) and equivariant GNN interatomic potentials, finding that neither domain has saturated with respect to model size, data, or compute.