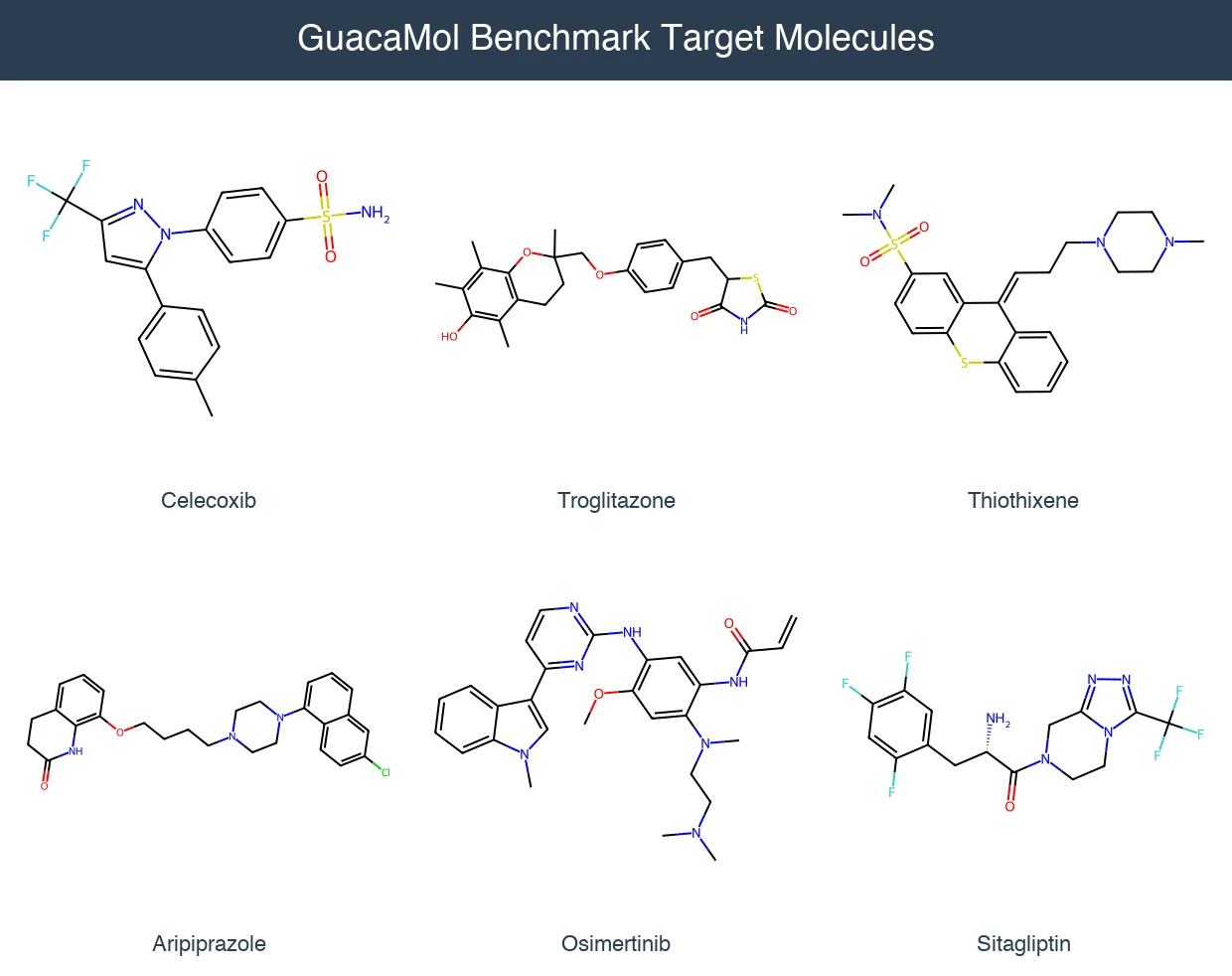
GuacaMol: Benchmarking Models for De Novo Molecular Design
GuacaMol provides an open-source benchmarking framework with 5 distribution-learning and 20 goal-directed tasks to standardize evaluation of de novo molecular design models.
GuacaMol provides an open-source benchmarking framework with 5 distribution-learning and 20 goal-directed tasks to standardize evaluation of de novo molecular design models.
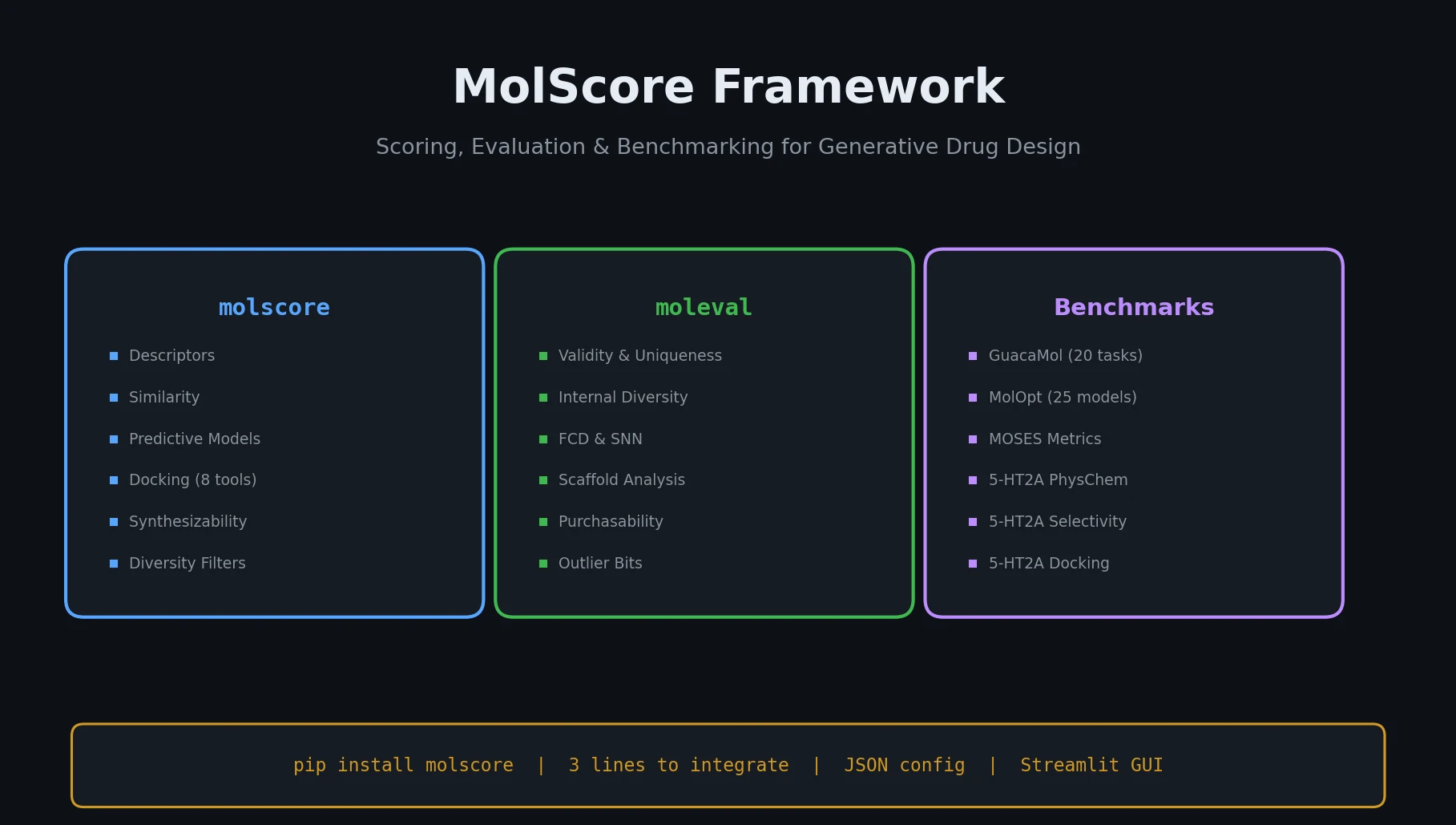
MolScore is an open-source framework that unifies scoring functions, evaluation metrics, and benchmarks for generative molecular design, with configurable objectives and GUI support.
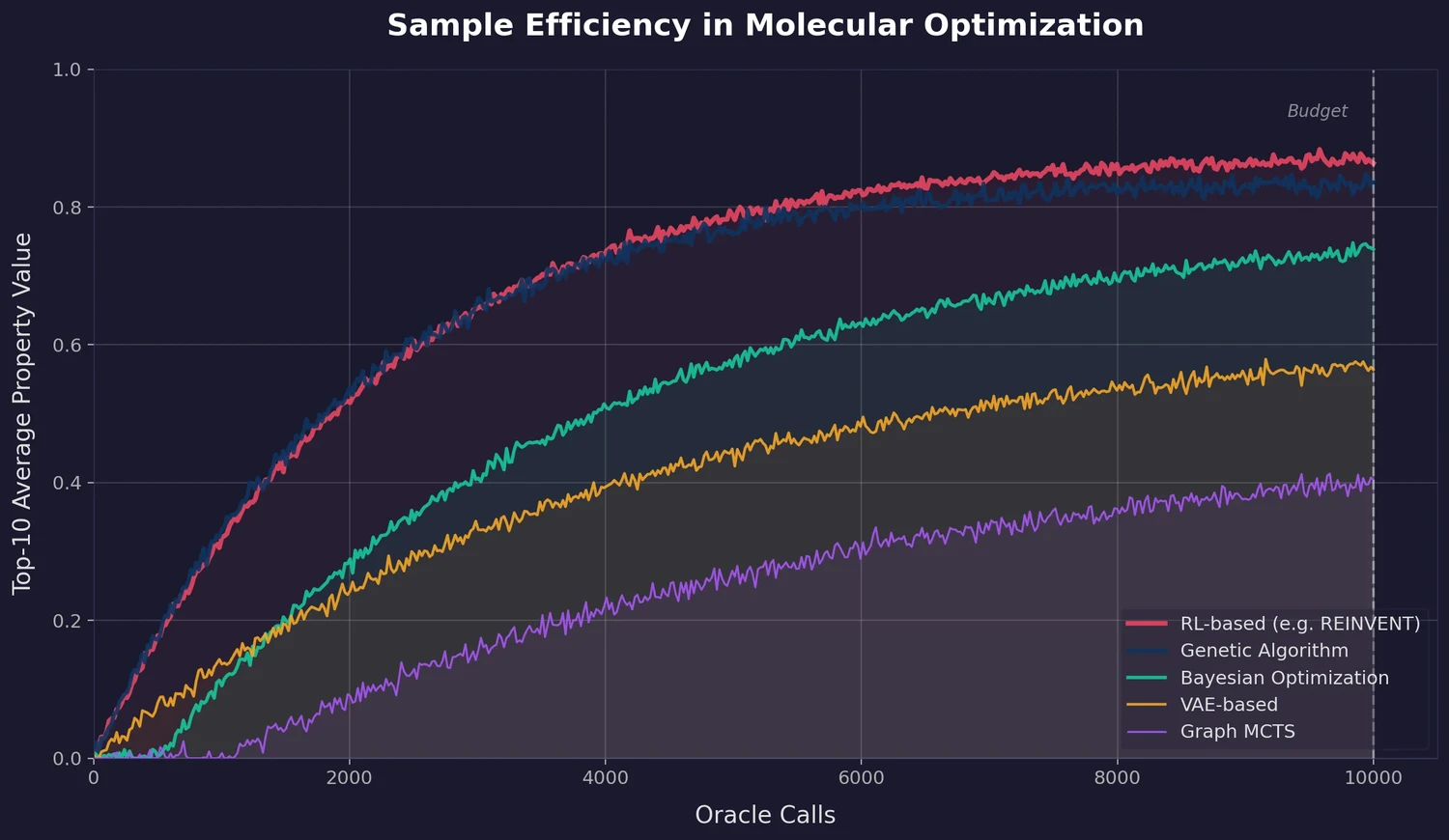
A large-scale benchmark of 25 molecular optimization methods on 23 oracles under constrained oracle budgets, showing that sample efficiency is a critical and often neglected dimension of evaluation.
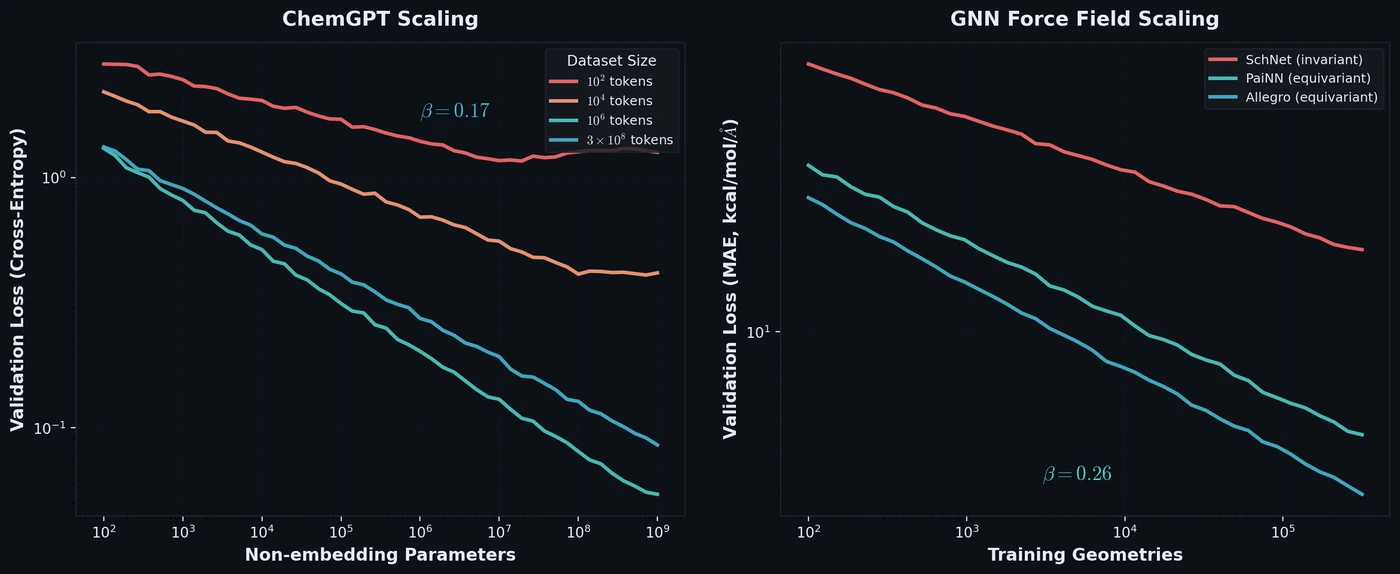
Frey et al. discover empirical power-law scaling relations for both chemical language models (ChemGPT, up to 1B parameters) and equivariant GNN interatomic potentials, finding that neither domain has saturated with respect to model size, data, or compute.
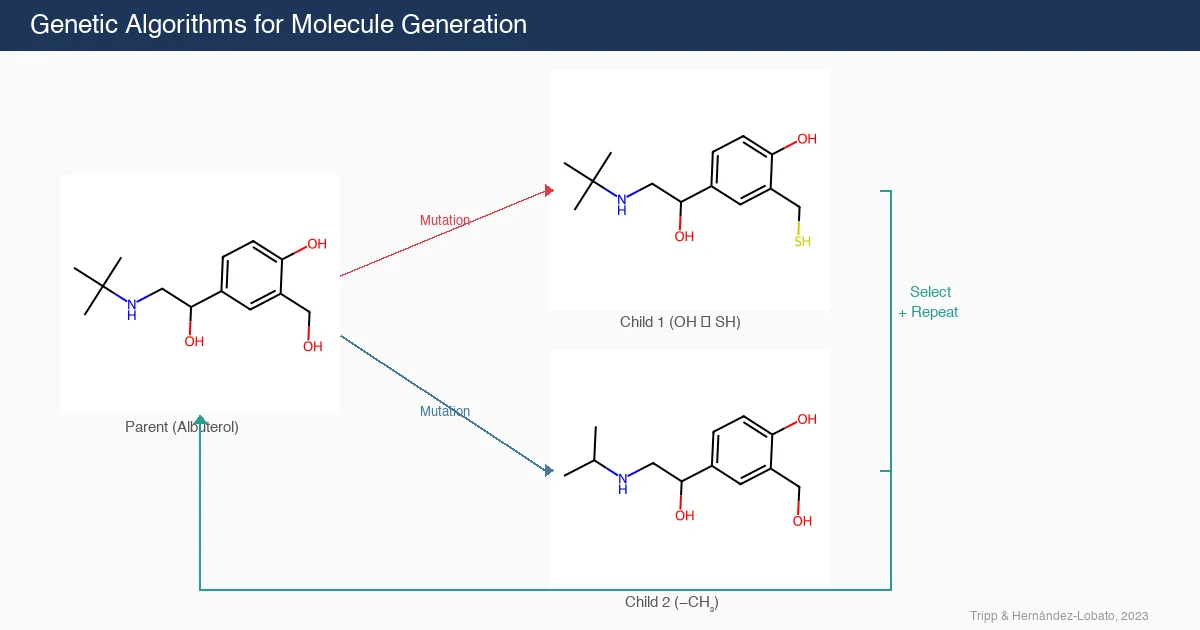
This position paper demonstrates that genetic algorithms (GAs) perform surprisingly well on molecular generation benchmarks, often outperforming complex deep learning methods. The authors propose the GA criterion: new molecule generation algorithms should demonstrate a clear advantage over GAs.
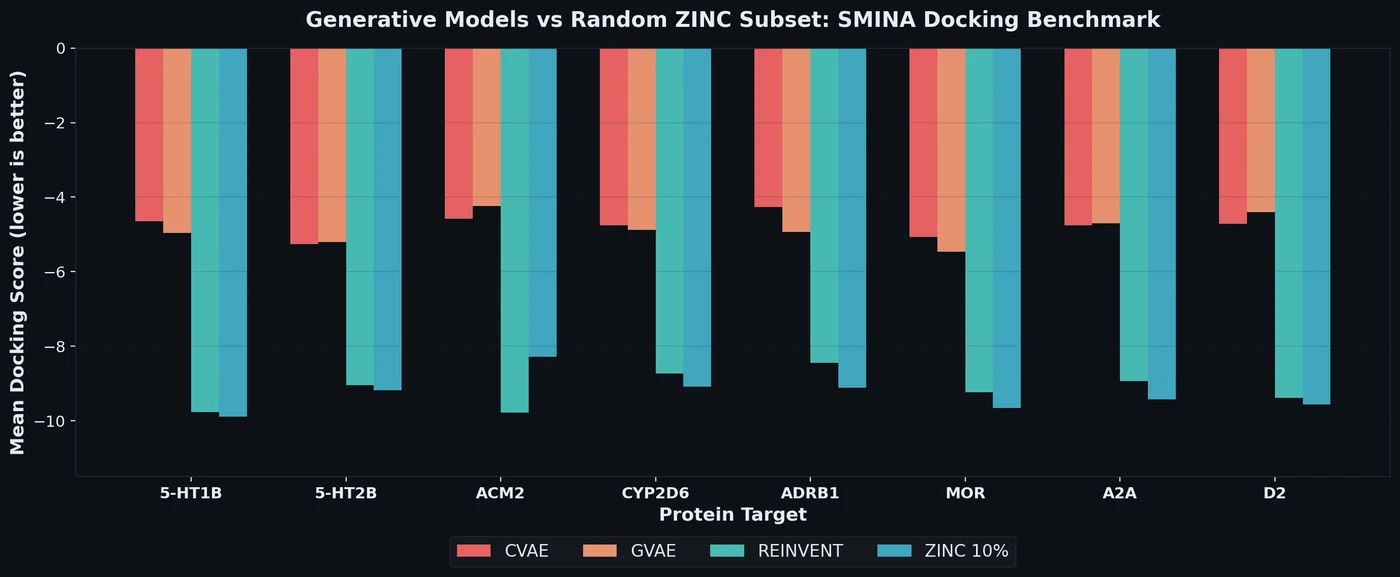
Proposes a benchmark for de novo drug design using SMINA docking scores across eight drug targets, revealing that popular generative models fail to outperform random ZINC subsets.
Tartarus introduces a modular suite of realistic molecular design benchmarks grounded in computational chemistry simulations. Benchmarking eight generative models reveals that no single algorithm dominates all tasks, and simple genetic algorithms often outperform deep generative models.
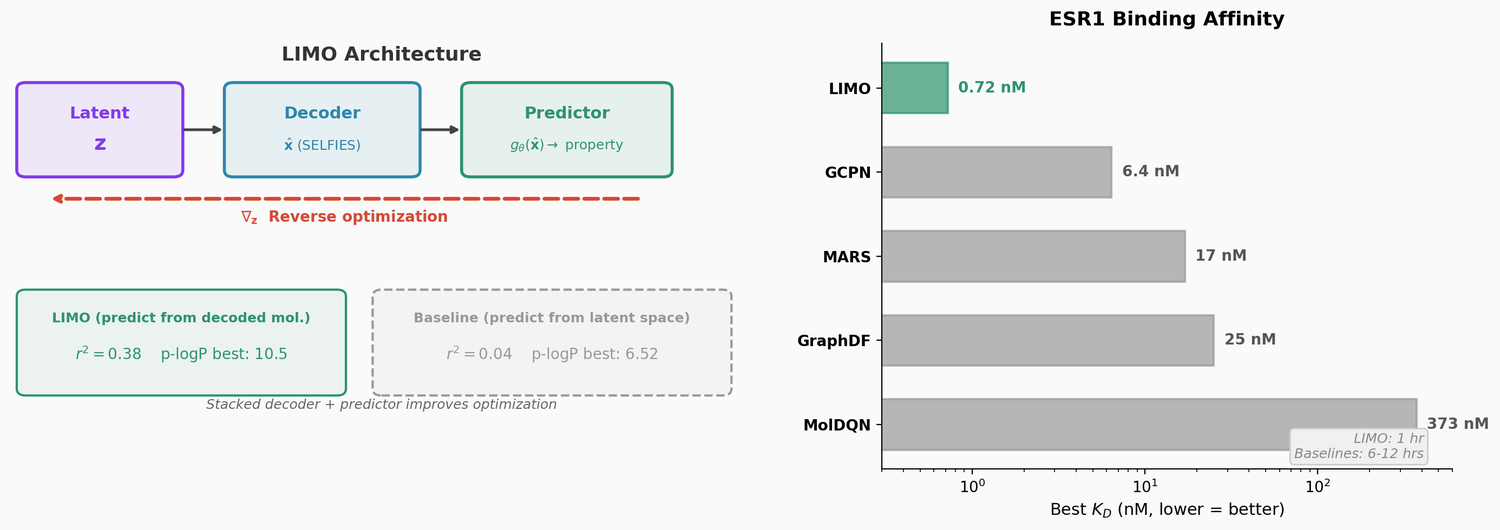
LIMO combines a SELFIES-based VAE with a novel stacked property predictor architecture (decoder output as predictor input) and gradient-based reverse optimization on the latent space. It is 6-8x faster than RL baselines and 12x faster than sampling methods while generating molecules with nanomolar binding affinities, including a predicted KD of 6e-14 M against the human estrogen receptor.
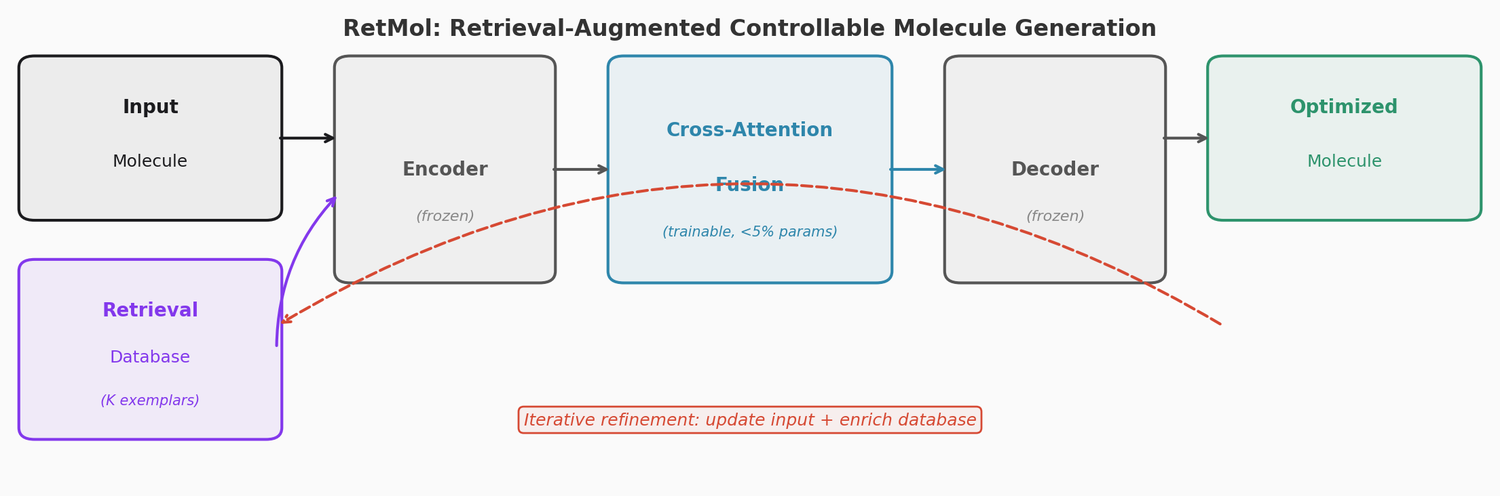
RetMol plugs a lightweight cross-attention retrieval module into a pre-trained Chemformer backbone to guide molecule generation toward multi-property design criteria. It requires no task-specific fine-tuning and works with as few as 23 exemplar molecules. It achieves 94.5% success on QED optimization, 96.9% on GSK3b/JNK3 dual inhibitor design, and 2.84 kcal/mol average binding affinity improvement on SARS-CoV-2 main protease inhibitor optimization.
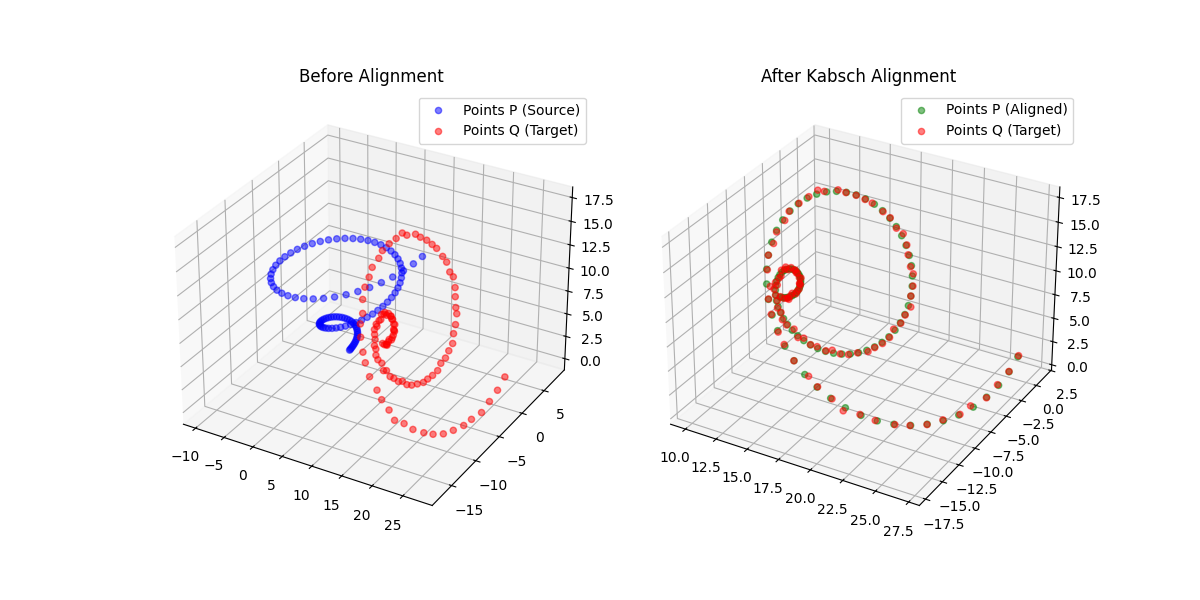
A differentiable point-set alignment library implementing N-dimensional Kabsch, Horn quaternion, and Umeyama scaling algorithms with per-point weights, batch dimensions, and custom autograd across NumPy, PyTorch, JAX, TensorFlow, and MLX.
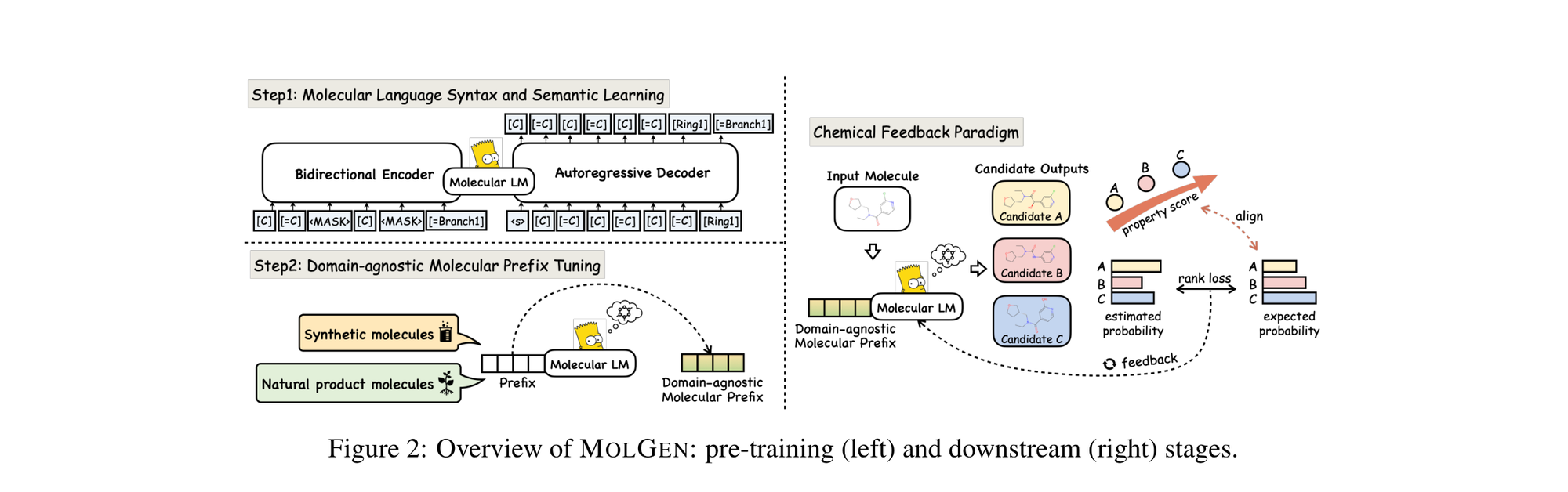
MolGen pre-trains on 100M+ SELFIES molecules, introduces domain-agnostic prefix tuning for cross-domain transfer, and applies a chemical feedback paradigm to reduce molecular hallucinations.
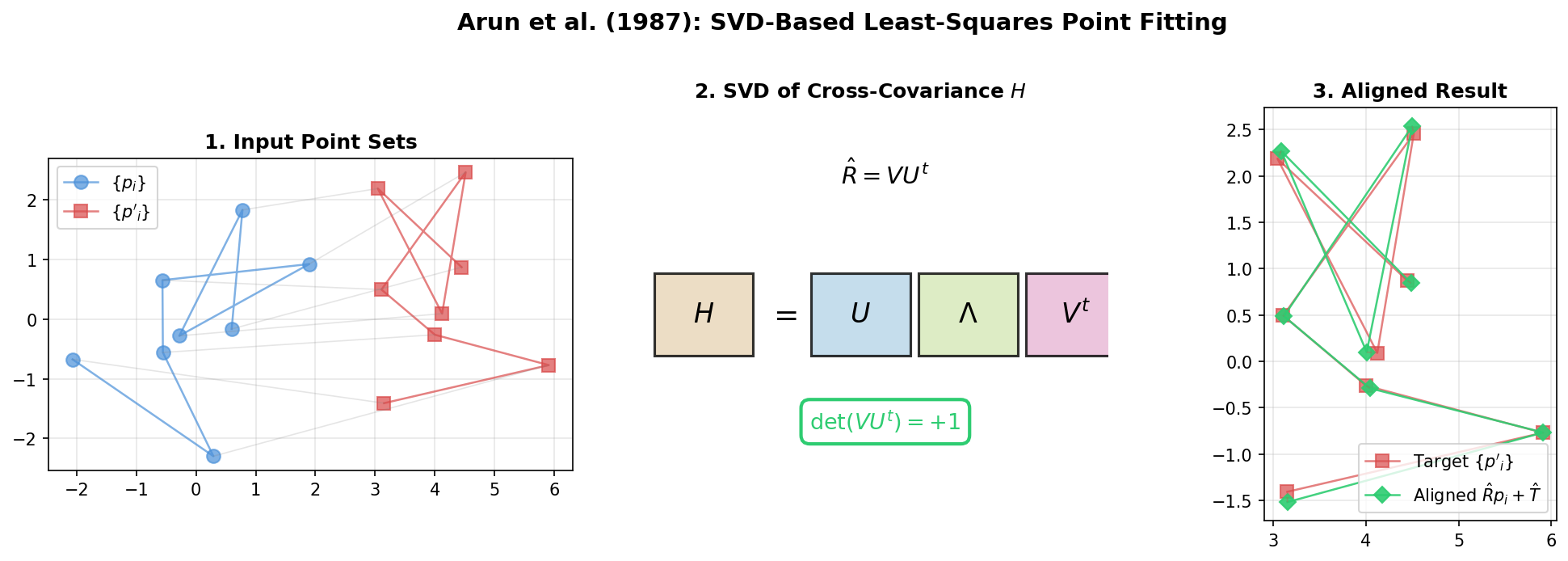
Presents a concise SVD-based algorithm for finding the optimal rotation and translation between two 3D point sets, with analysis of the degenerate reflection case that Umeyama later corrected.