Coscientist: Autonomous Chemistry with LLM Agents
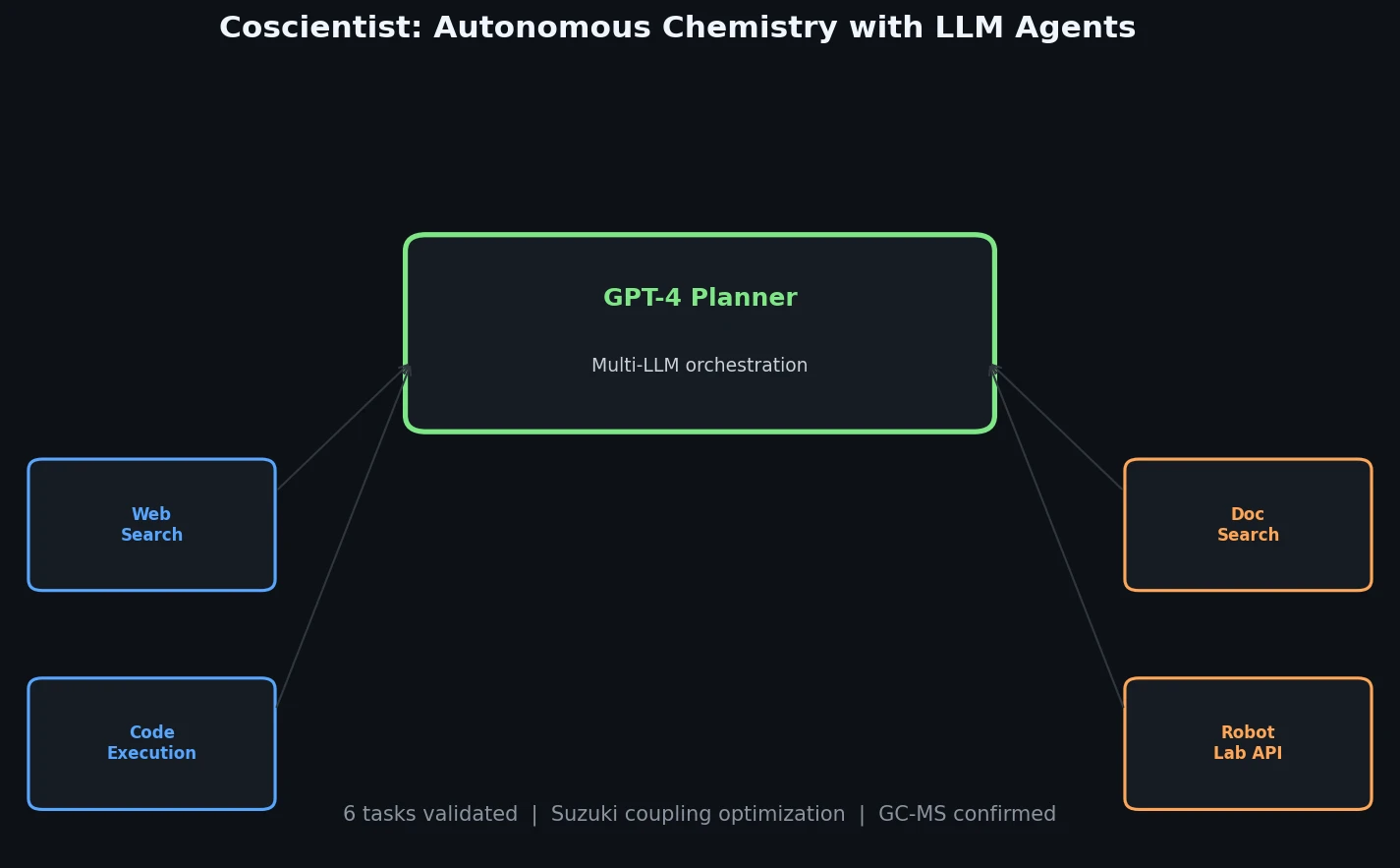
Introduces Coscientist, a GPT-4-driven AI system that autonomously designs and executes chemical experiments using web search, code execution, and robotic lab automation.
Introduces Coscientist, a GPT-4-driven AI system that autonomously designs and executes chemical experiments using web search, code execution, and robotic lab automation.
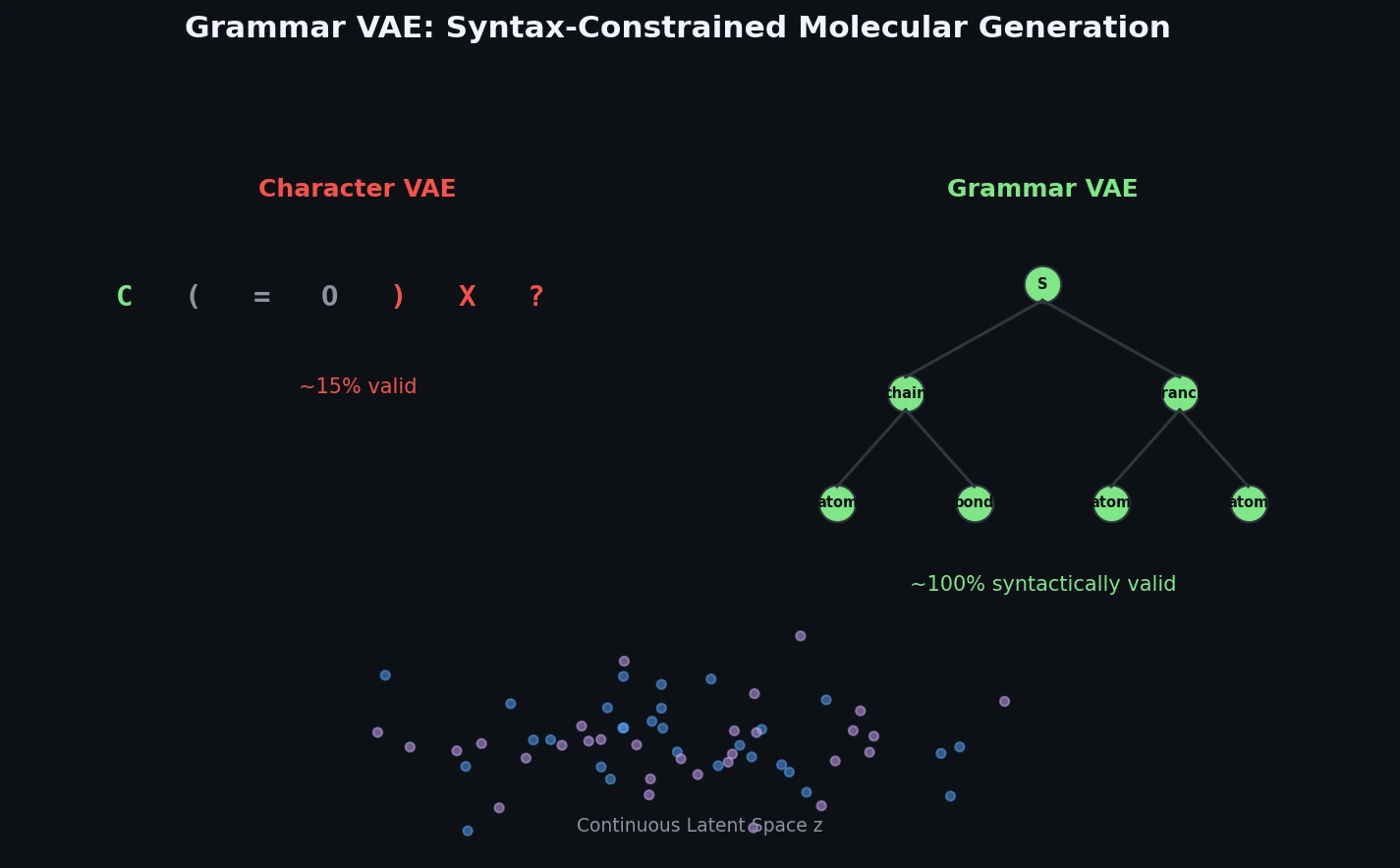
The Grammar VAE replaces character-level decoding with context-free grammar production rules, using a stack-based masking mechanism to guarantee that all generated SMILES strings are syntactically valid. Applied to molecular optimization and symbolic regression, it learns smoother latent spaces and finds better molecules than character-level baselines.
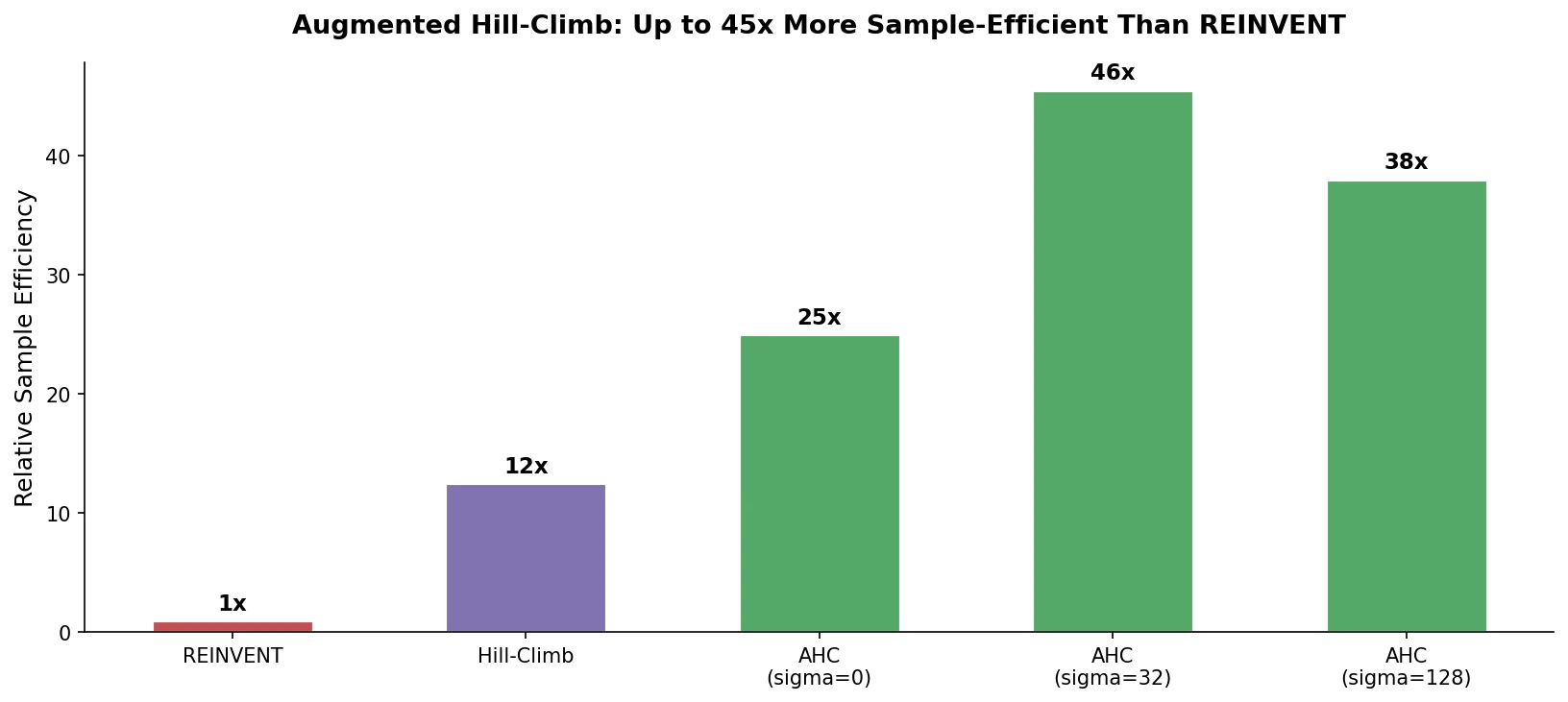
Proposes Augmented Hill-Climb, a hybrid RL strategy for SMILES-based generative models that improves sample efficiency ~45-fold over REINVENT by filtering low-scoring molecules from the loss computation, with diversity filters to prevent mode collapse.
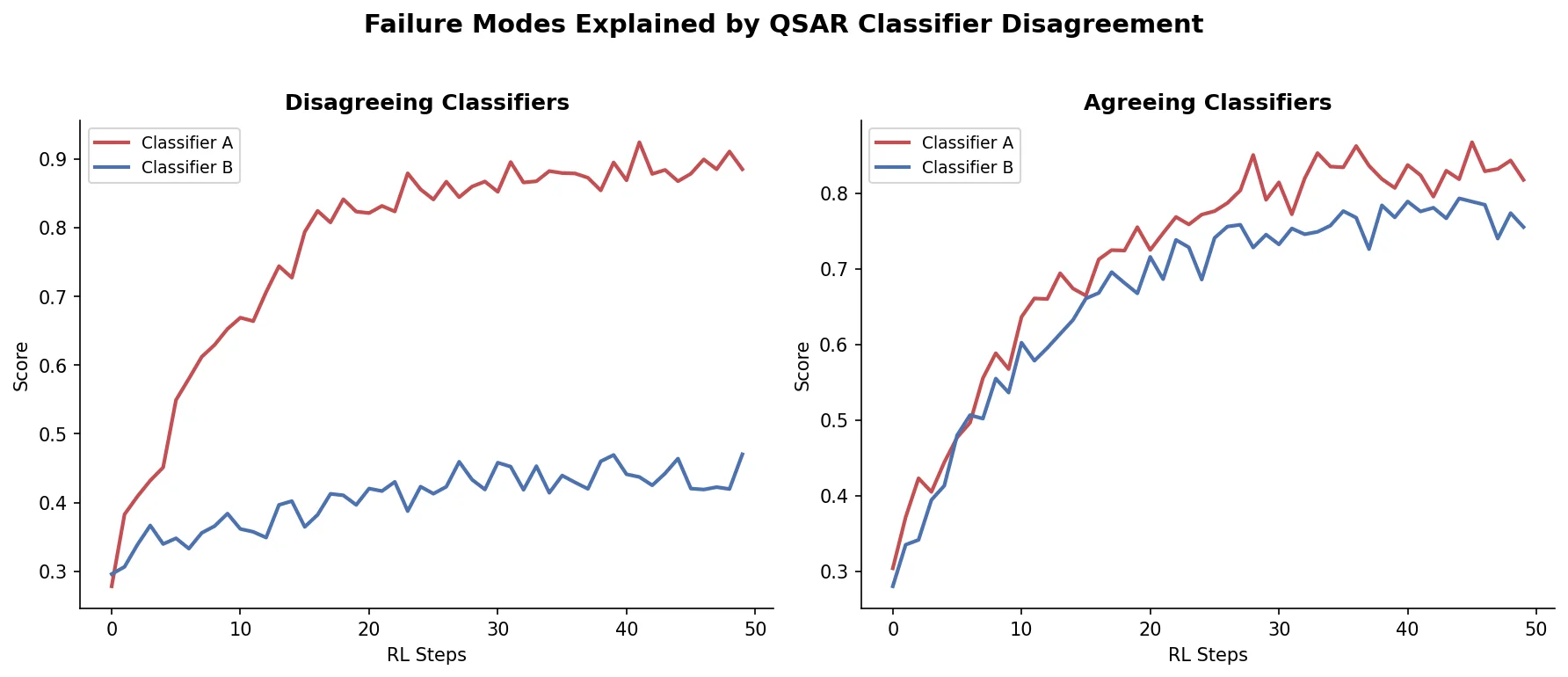
Shows that divergence between optimization and control scores during goal-directed molecular generation is explained by pre-existing disagreement among QSAR models on the training distribution, not by algorithmic exploitation of model-specific biases.
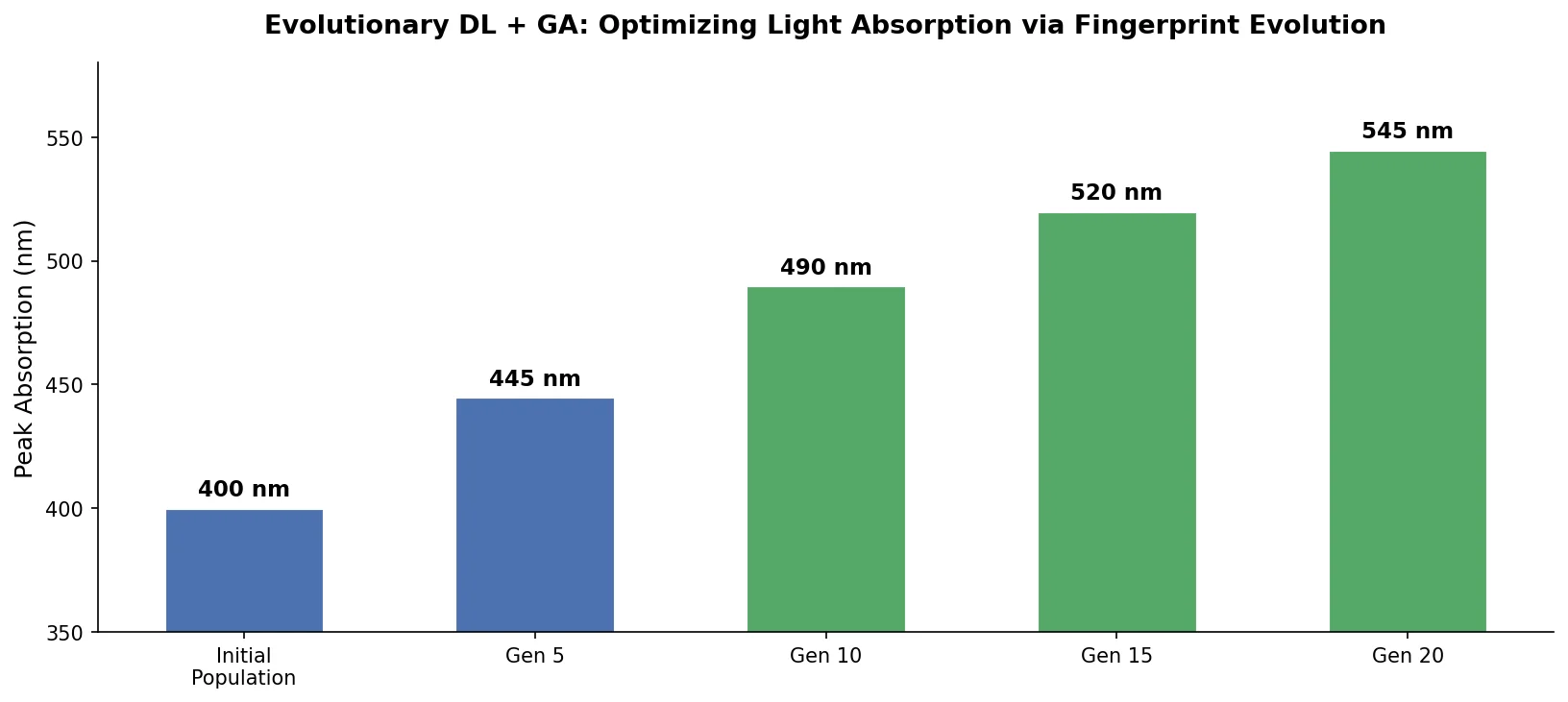
An evolutionary molecular design framework that evolves ECFP fingerprint vectors using a genetic algorithm, reconstructs valid SMILES via an RNN decoder, and evaluates fitness with a DNN property predictor.
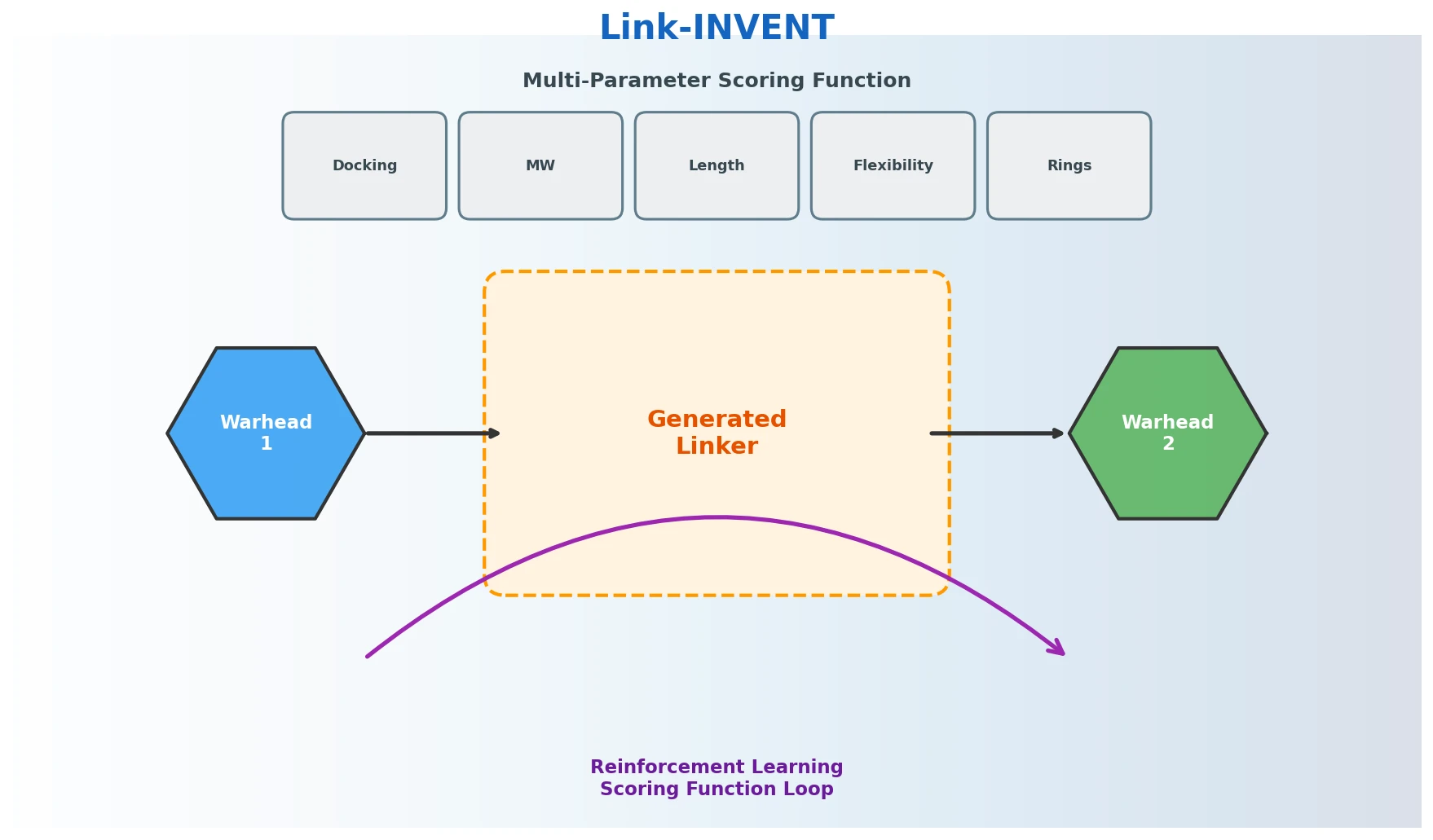
Link-INVENT is an RNN-based generative model for molecular linker design that uses reinforcement learning with a flexible scoring function, demonstrated on fragment linking, scaffold hopping, and PROTAC design.
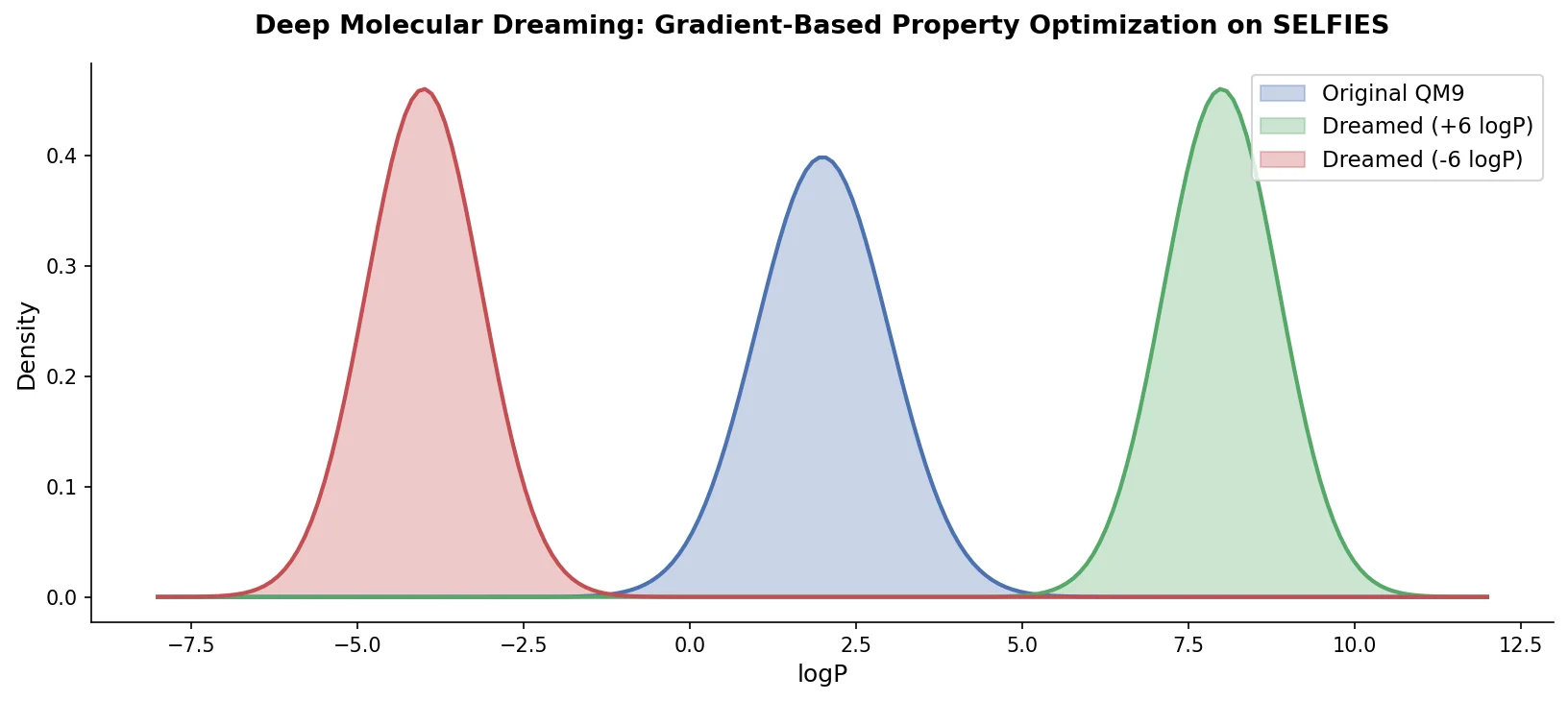
PASITHEA adapts deep dreaming from computer vision to molecular design, directly optimizing SELFIES-encoded molecules for target chemical properties via gradient-based inversion of a trained regression network.
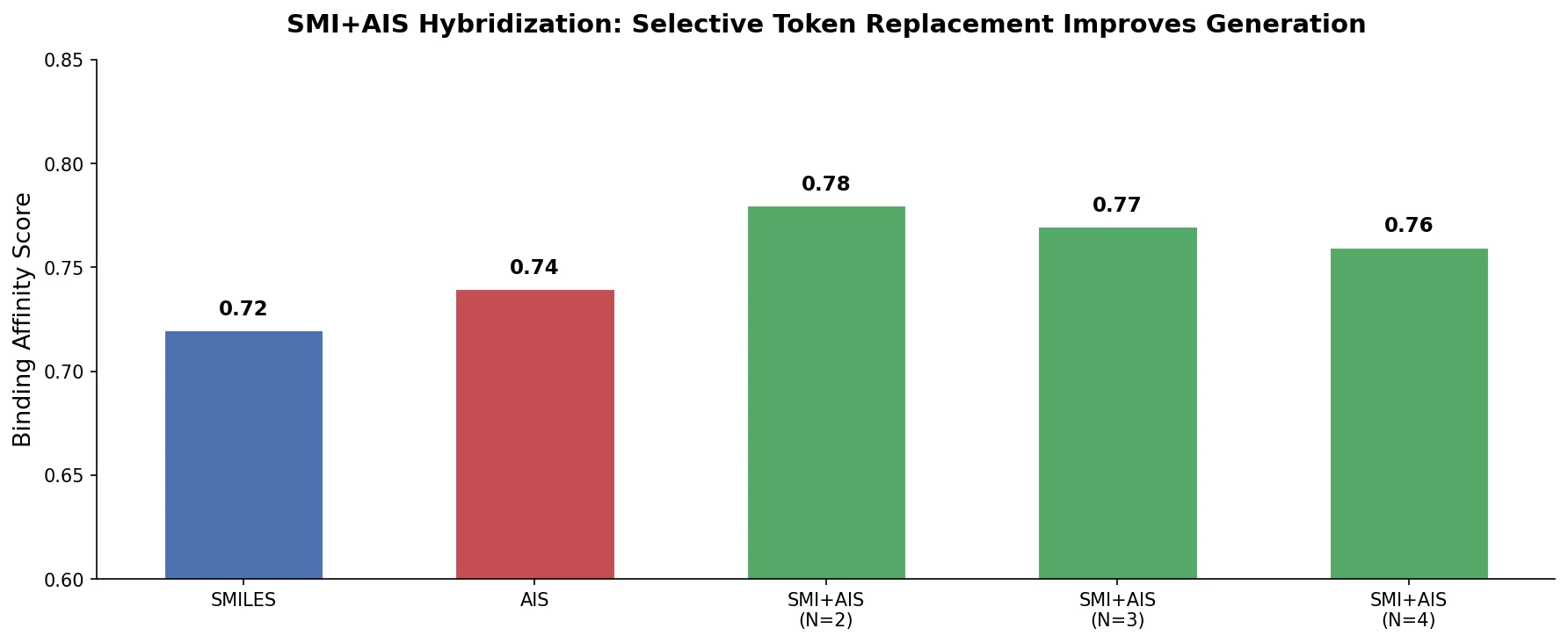
Proposes SMI+AIS, a hybrid molecular representation combining standard SMILES tokens with chemical-environment-aware Atom-In-SMILES tokens, demonstrating improved molecular generation for drug design targets.
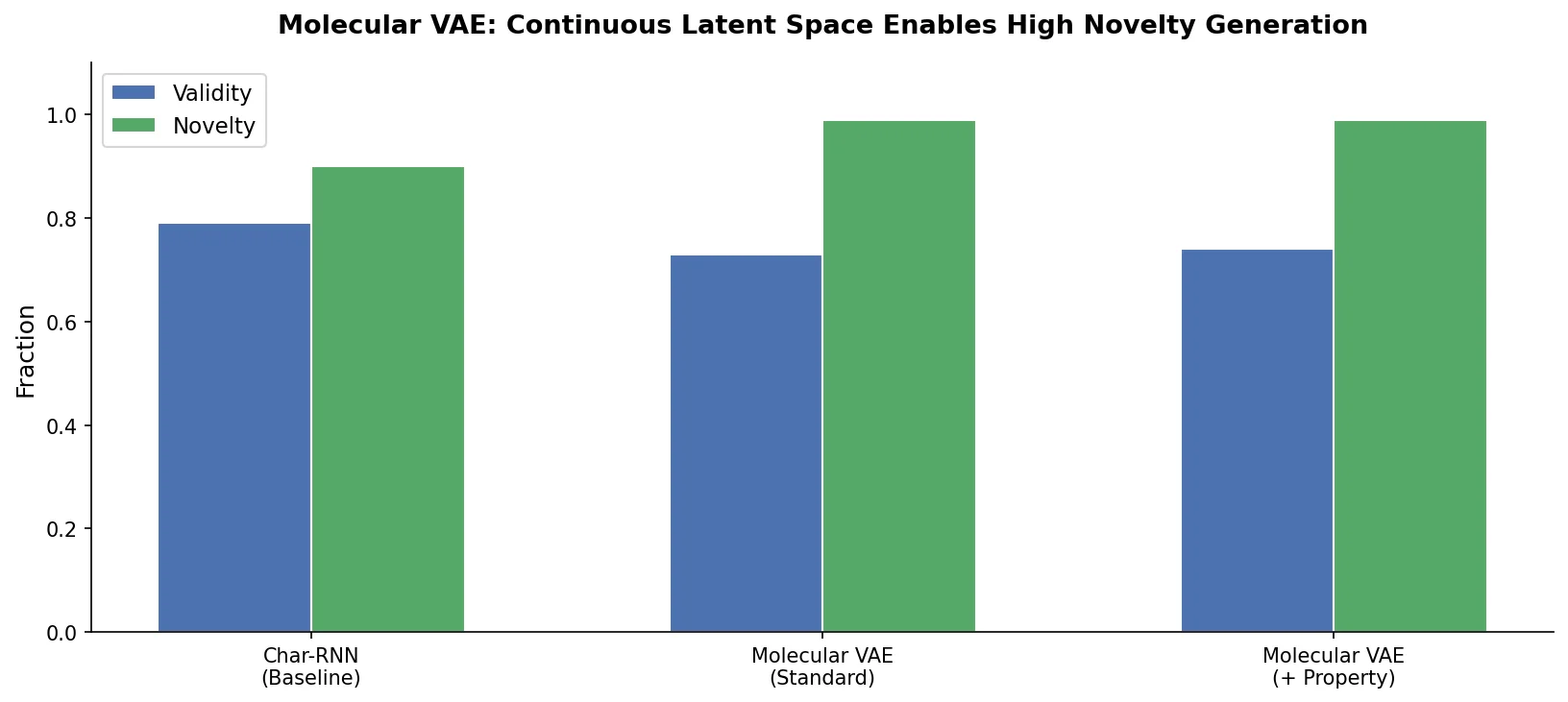
This foundational paper introduces a variational autoencoder (VAE) that encodes SMILES strings into a continuous latent space, allowing gradient-based optimization of molecular properties. Joint training with a property predictor organizes the latent space by chemical properties, and Bayesian optimization over the latent surface discovers drug-like molecules with improved QED and synthetic accessibility.
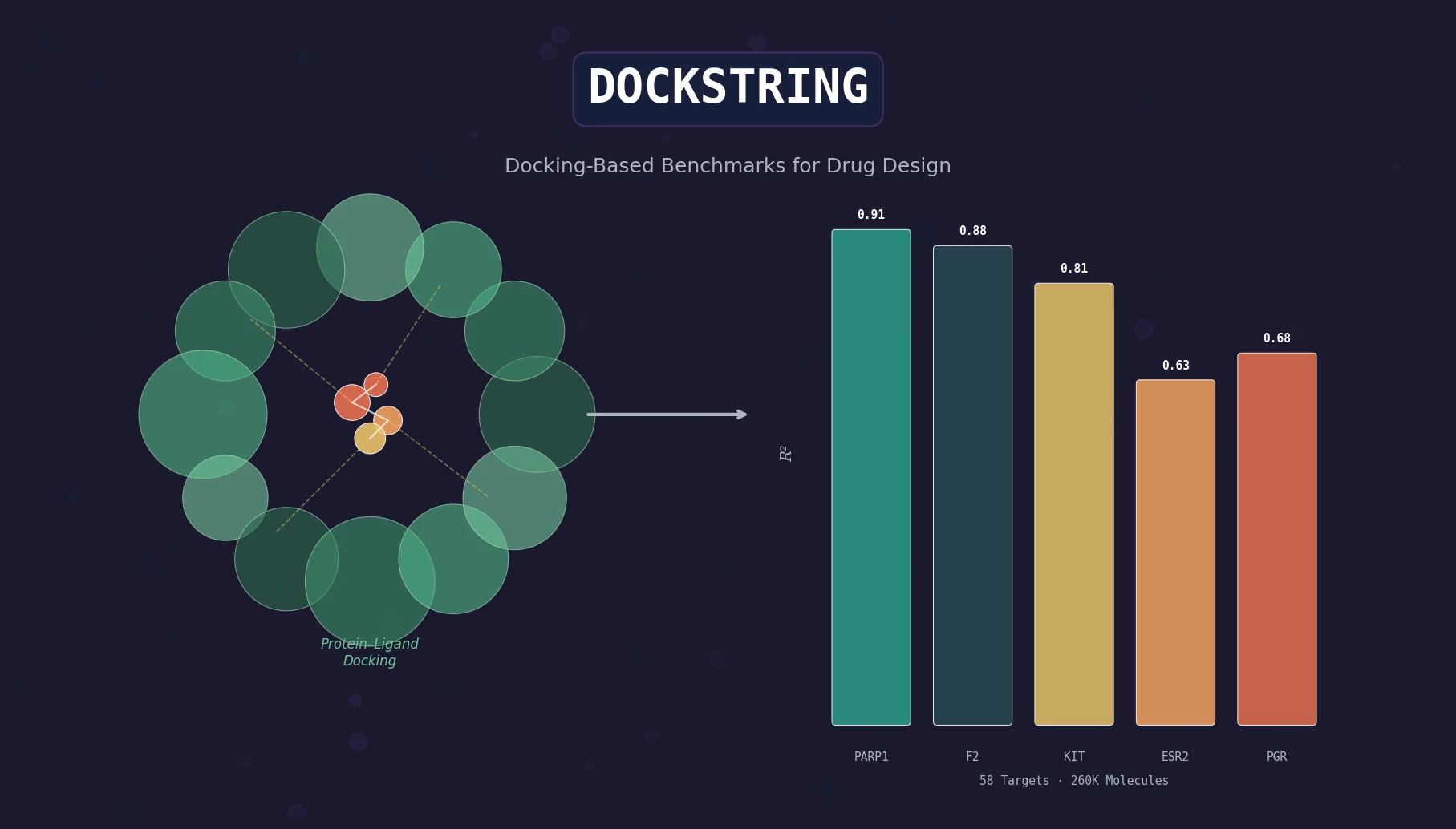
DOCKSTRING bundles an AutoDock Vina wrapper, a 260K-molecule docking dataset across 58 protein targets, and pharmaceutically relevant benchmarks for regression, virtual screening, and de novo design.
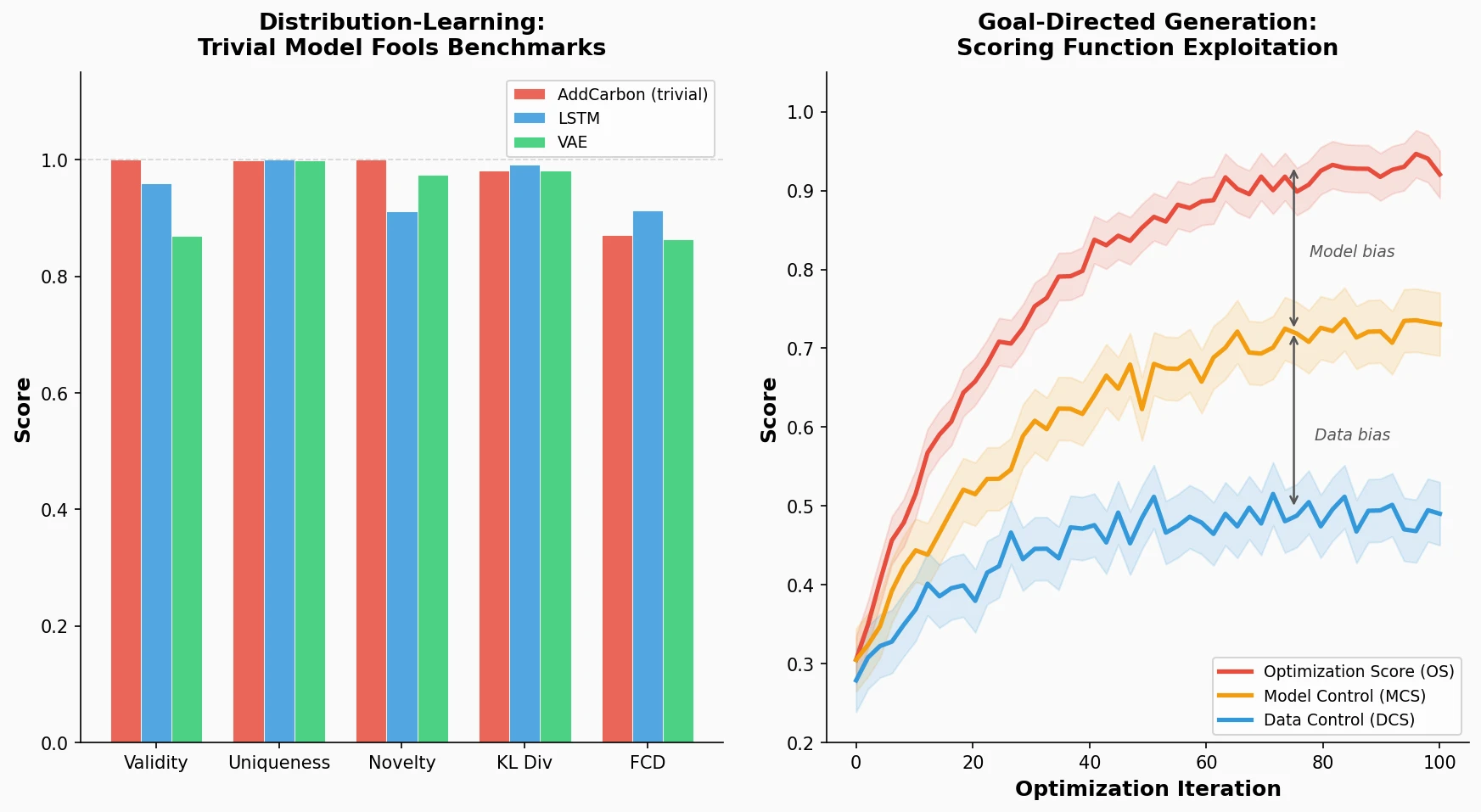
Identifies failure modes in molecular generative models, showing that trivial edits fool distribution-learning benchmarks and that ML-based scoring functions introduce exploitable model-specific and data-specific biases during goal-directed optimization.
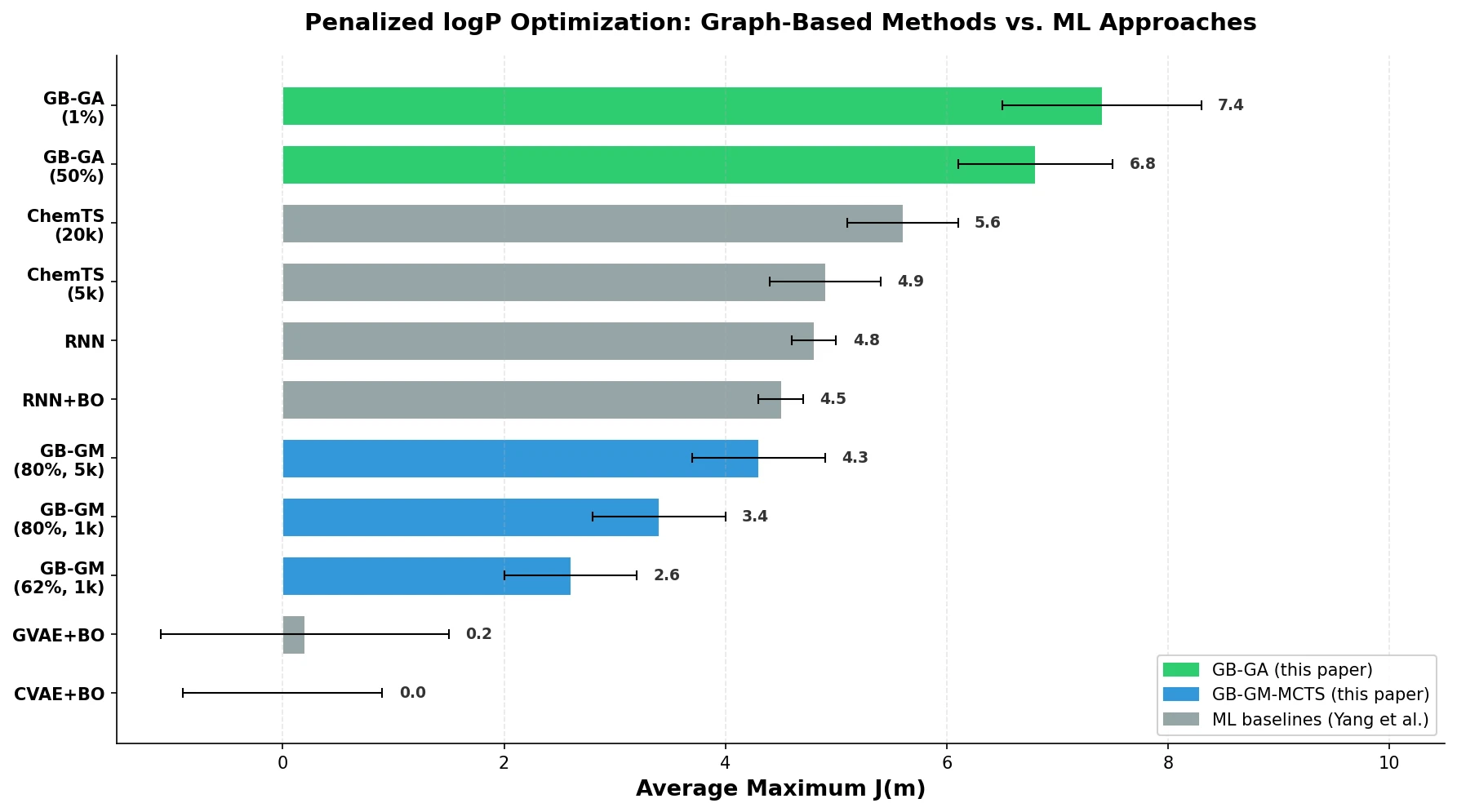
A graph-based genetic algorithm (GB-GA) and a graph-based generative model with Monte Carlo tree search (GB-GM-MCTS) for molecular optimization that match or outperform ML-based generative approaches while being orders of magnitude faster.