Smirk: Complete Tokenization for Molecular Models
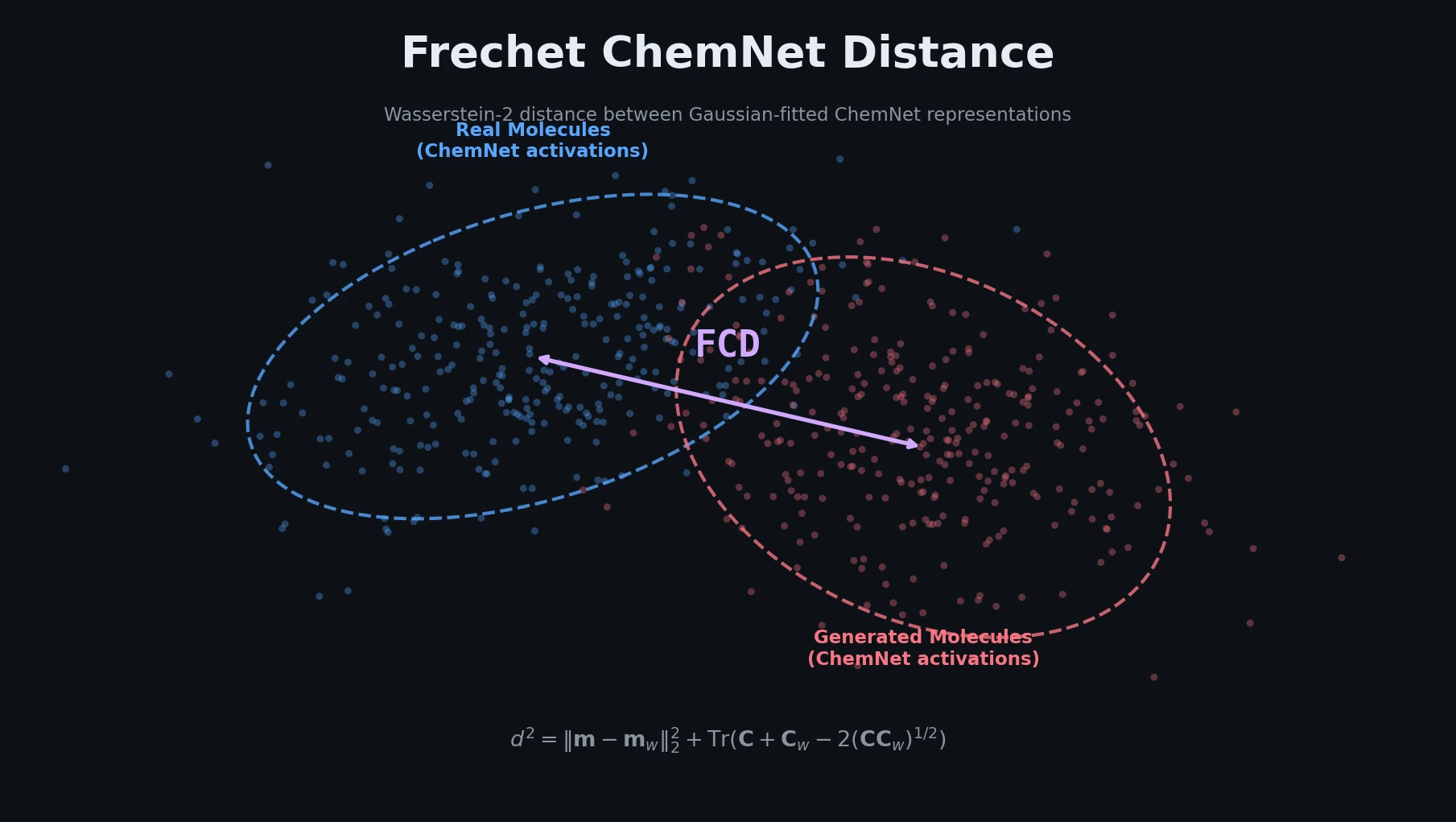
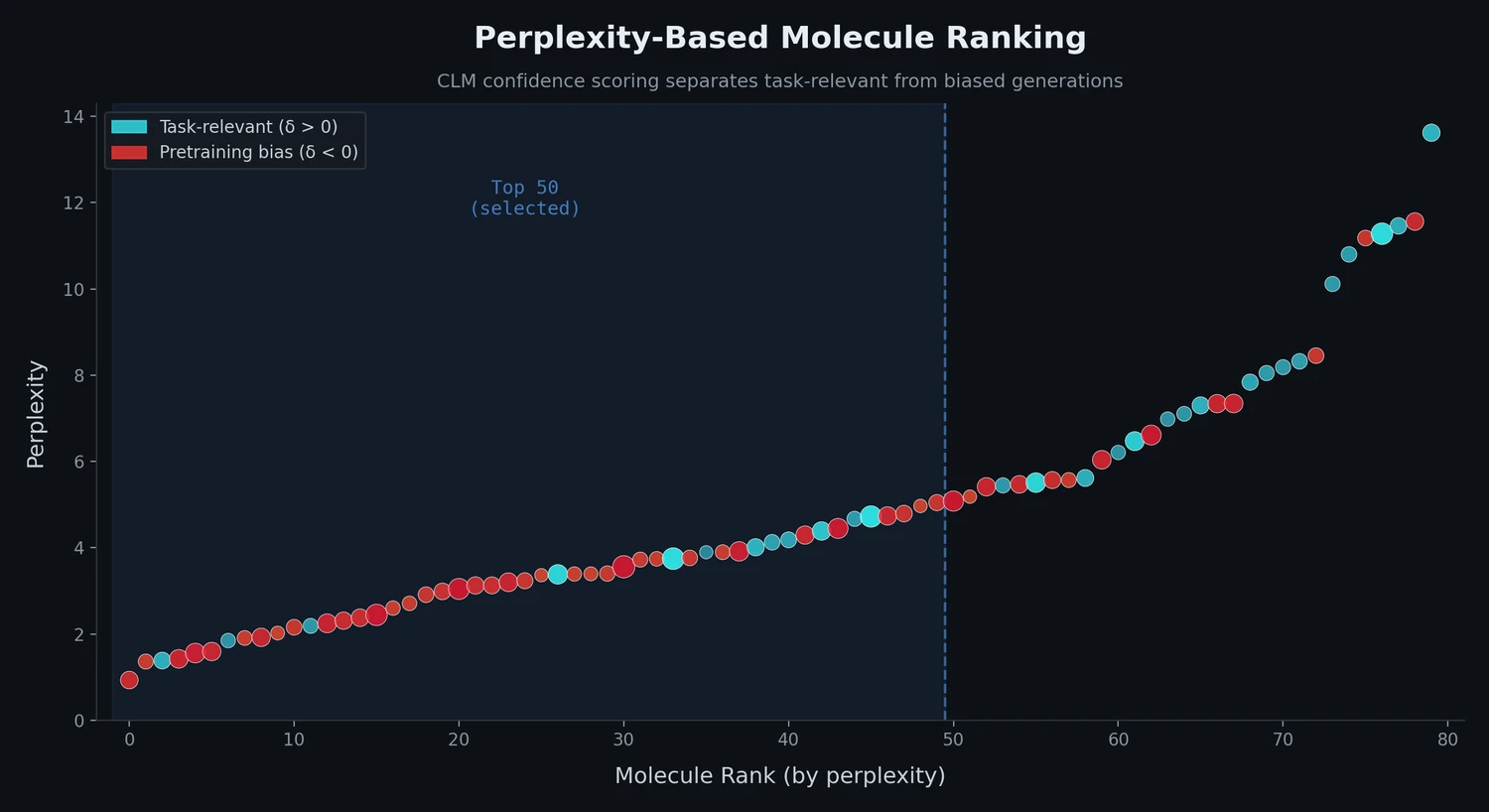
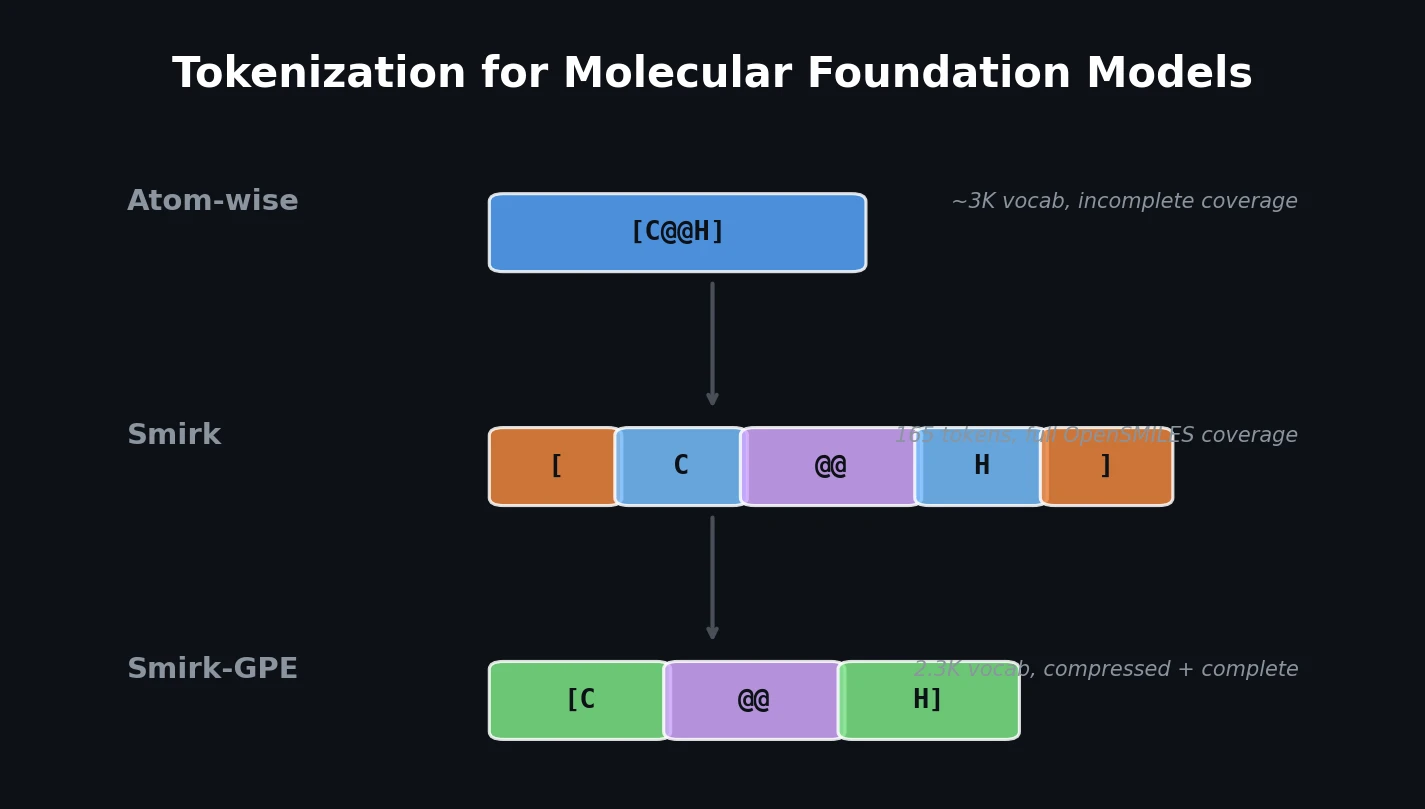
Introduces Smirk and Smirk-GPE tokenizers that fully cover the OpenSMILES specification, proposes n-gram language models as low-cost proxies for evaluating tokenizer quality, and benchmarks 34 tokenizers across intrinsic and extrinsic metrics.