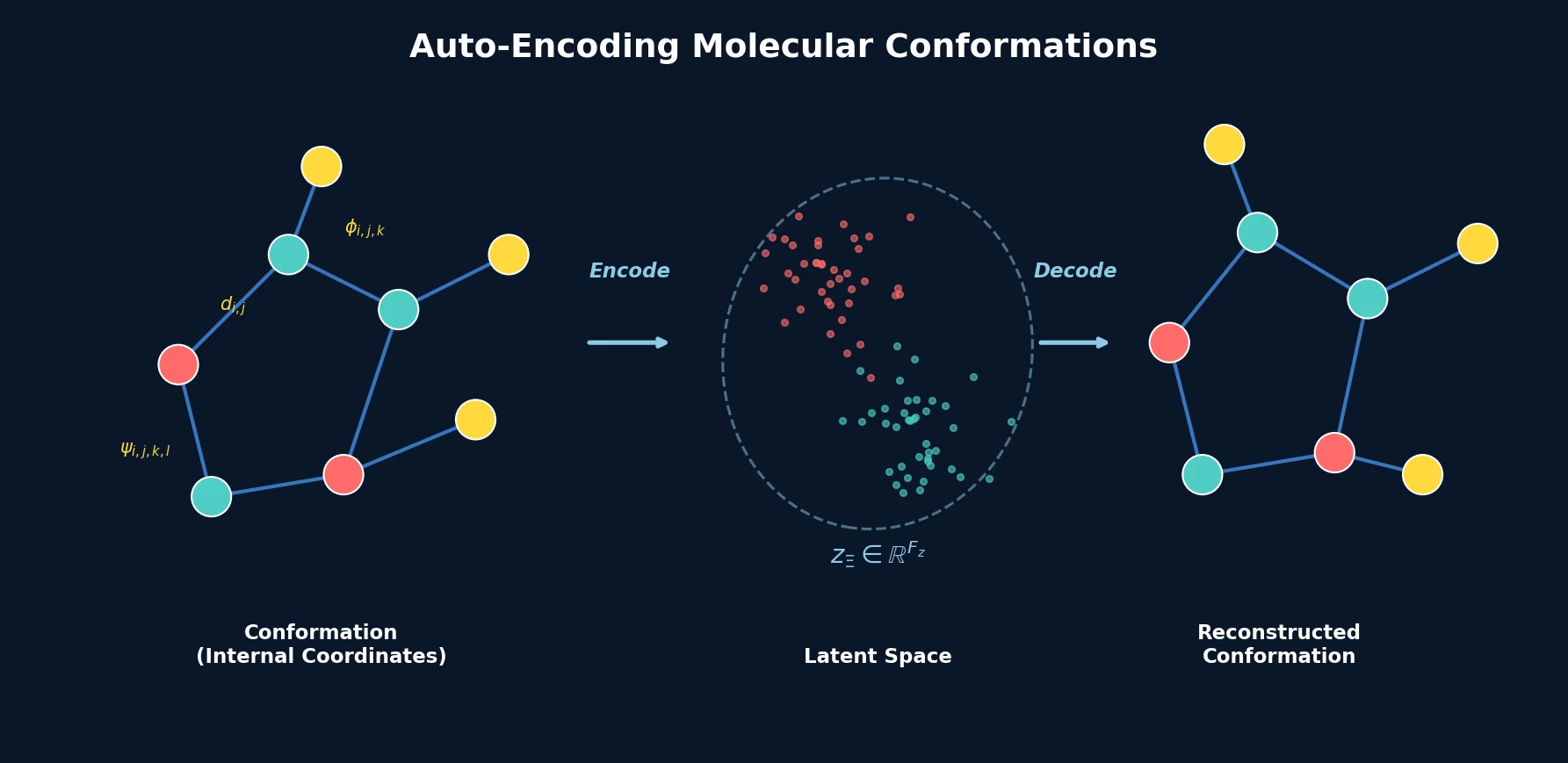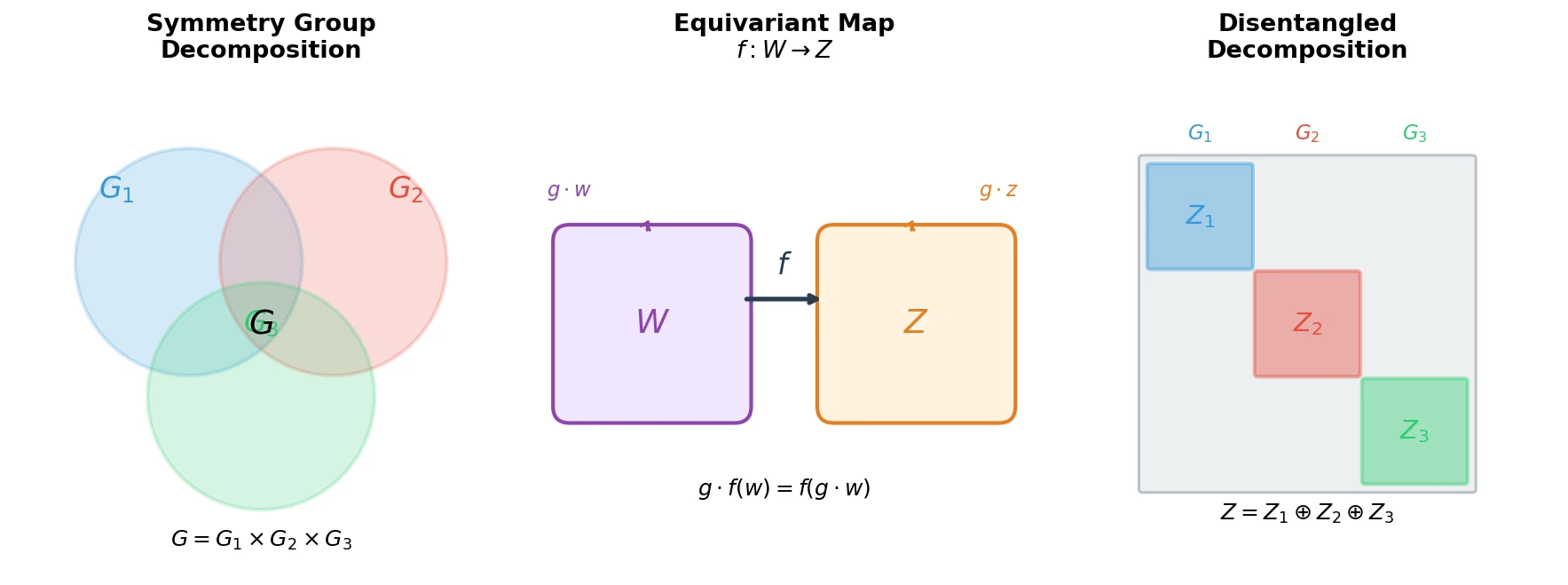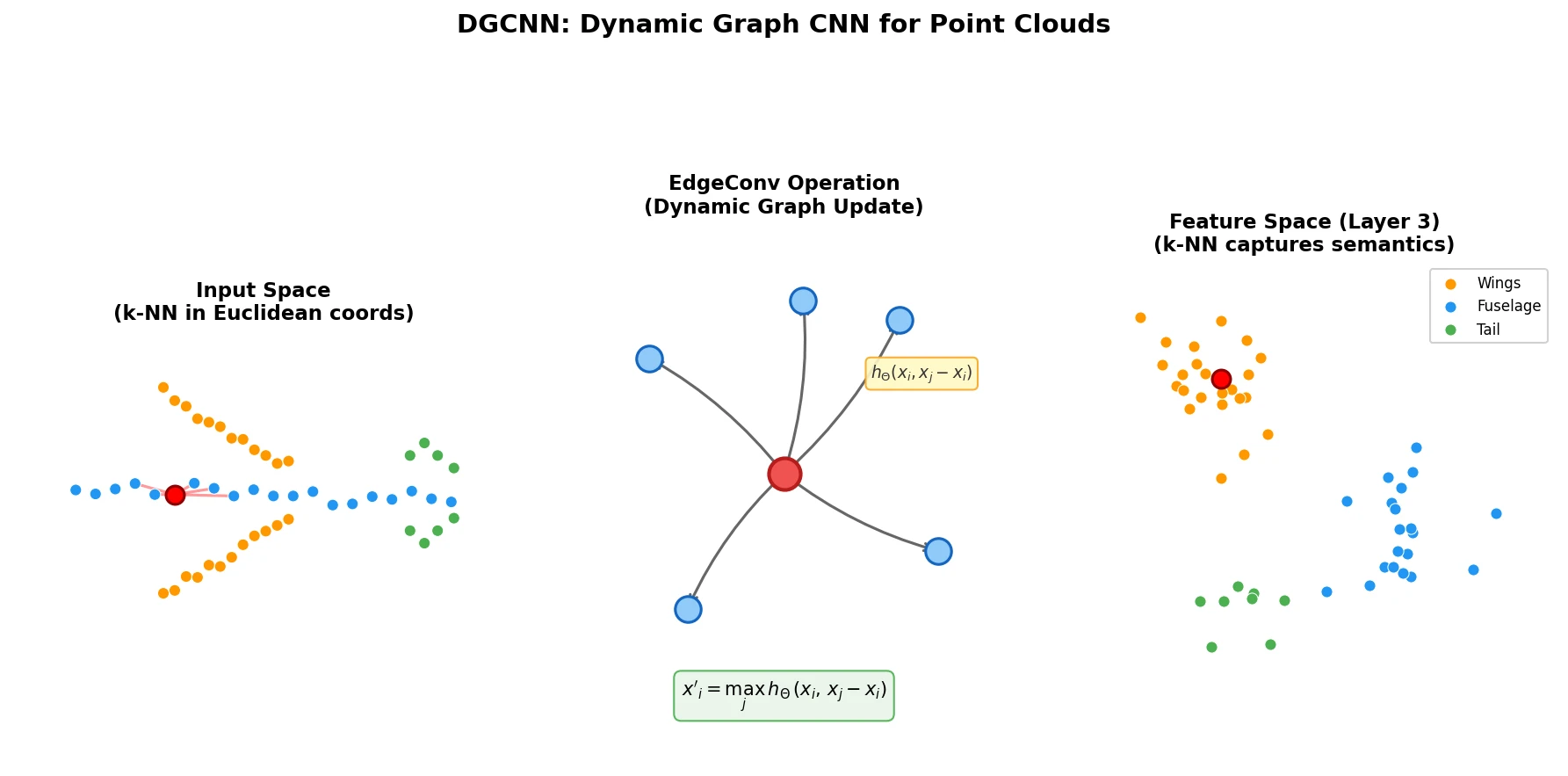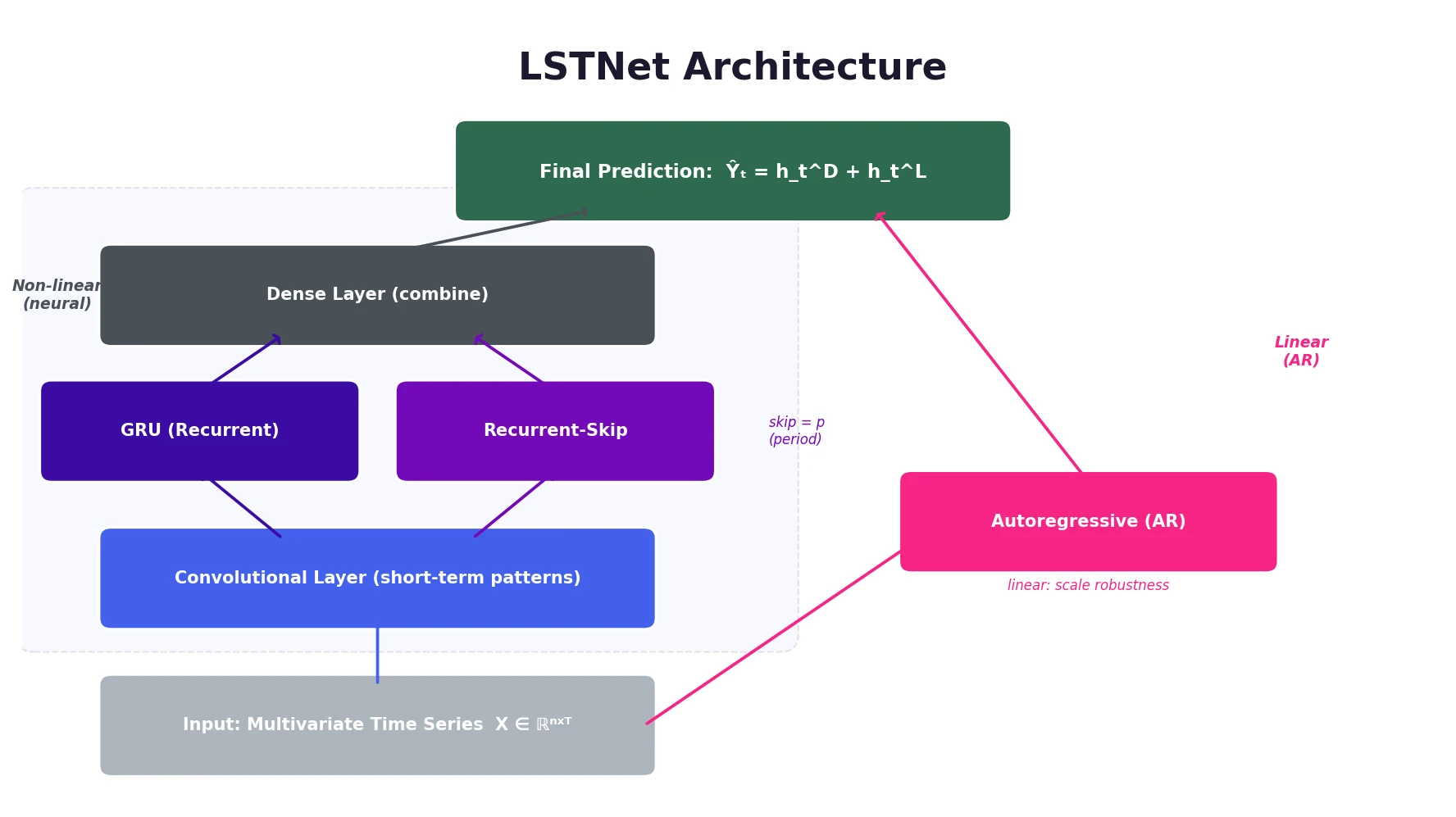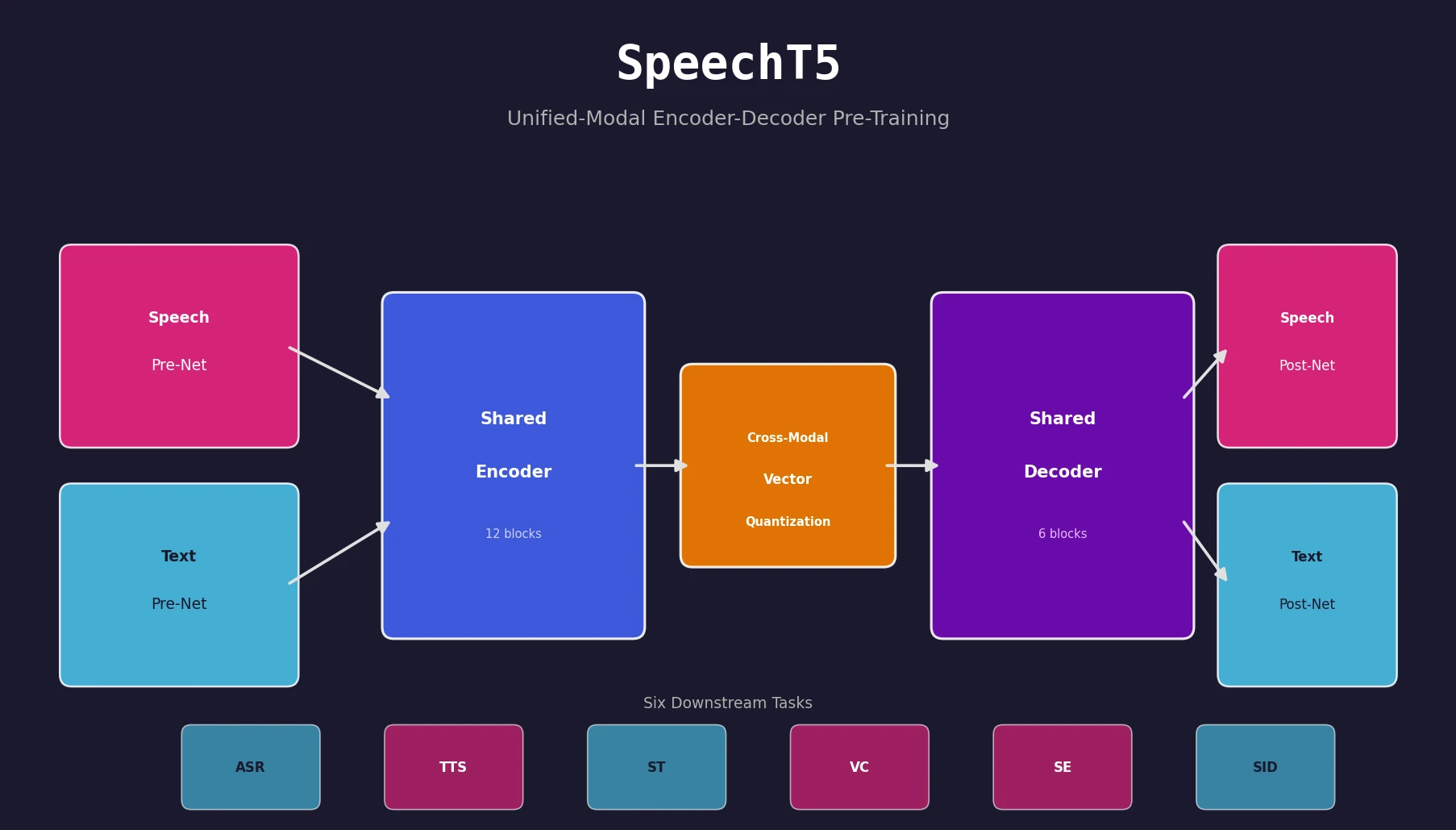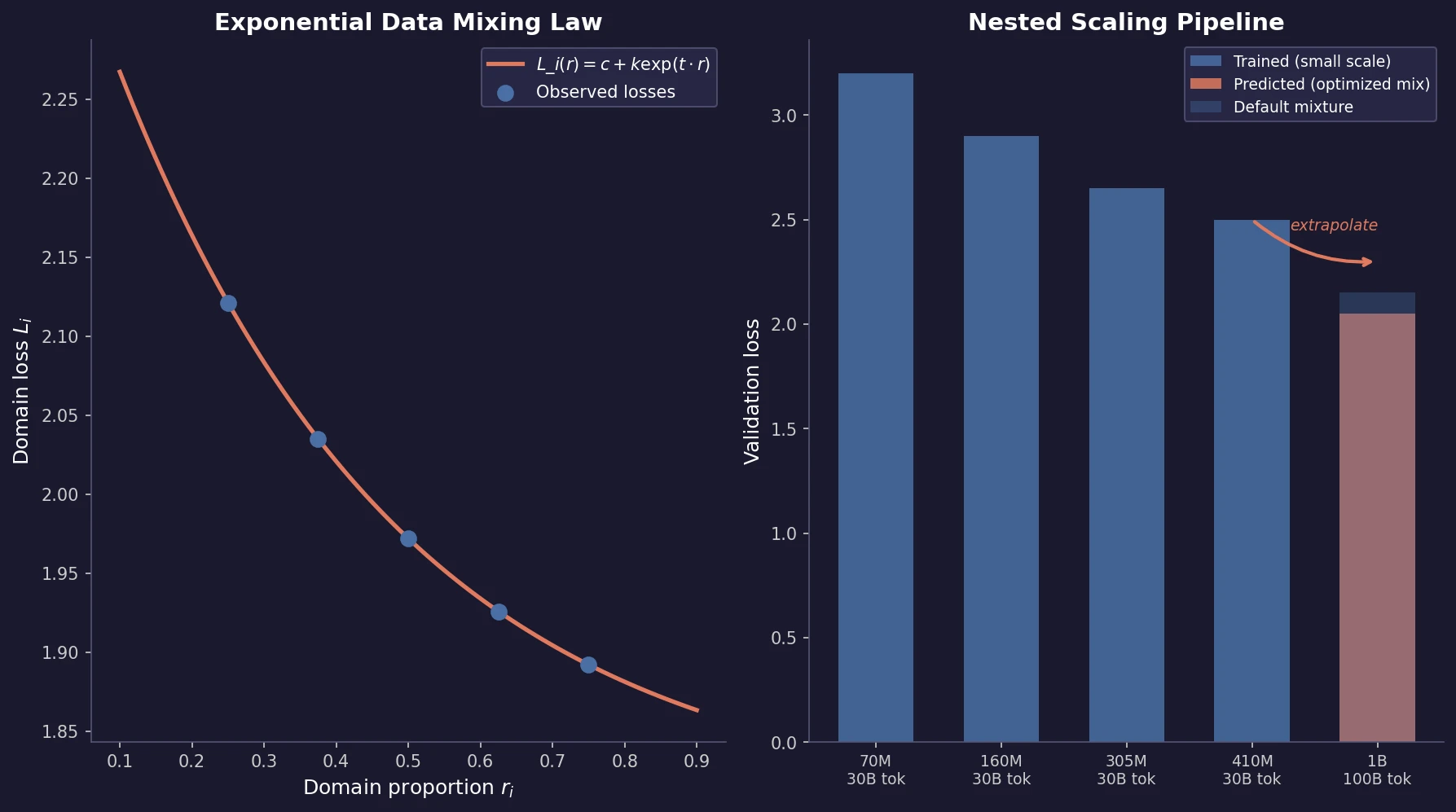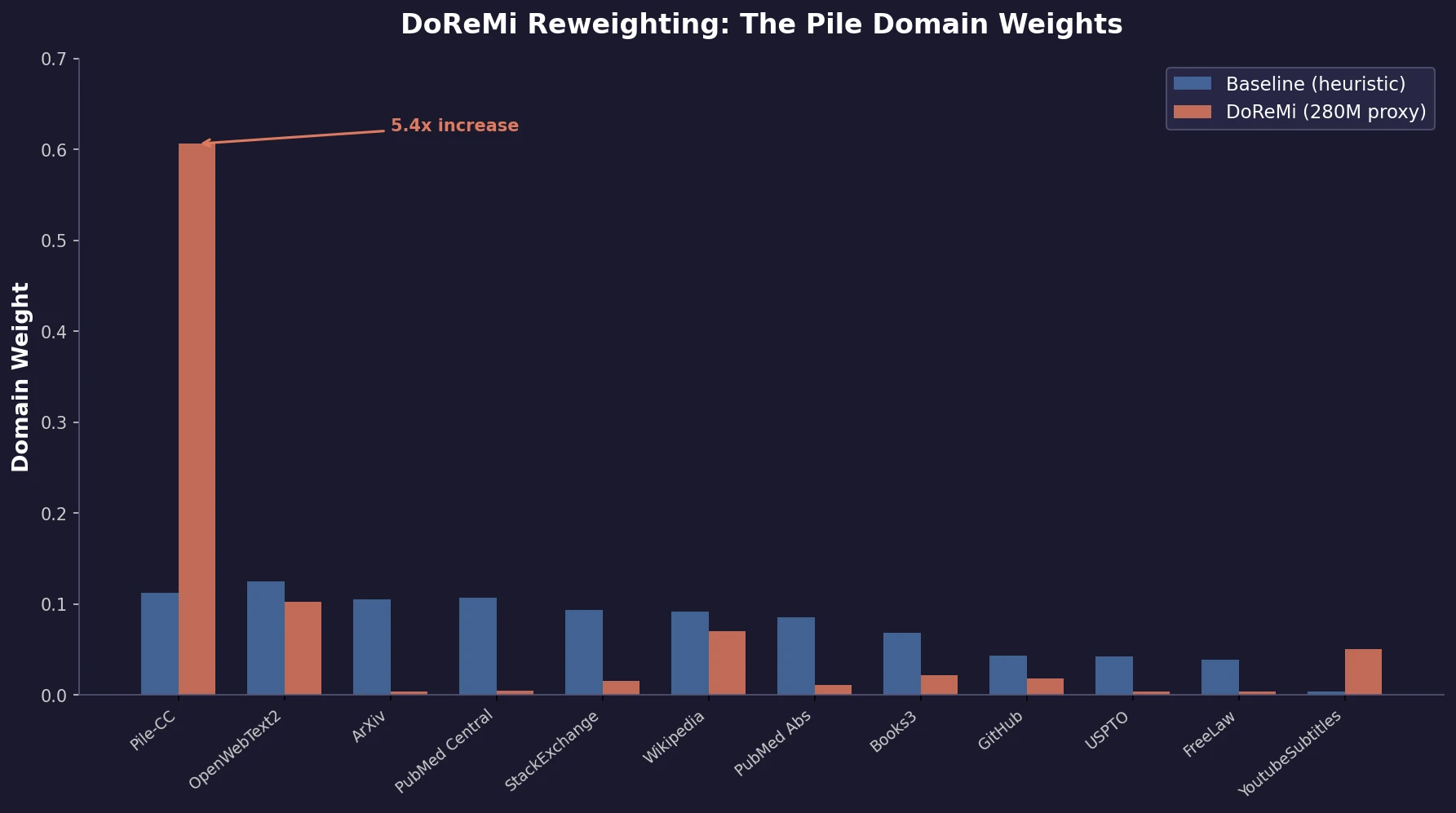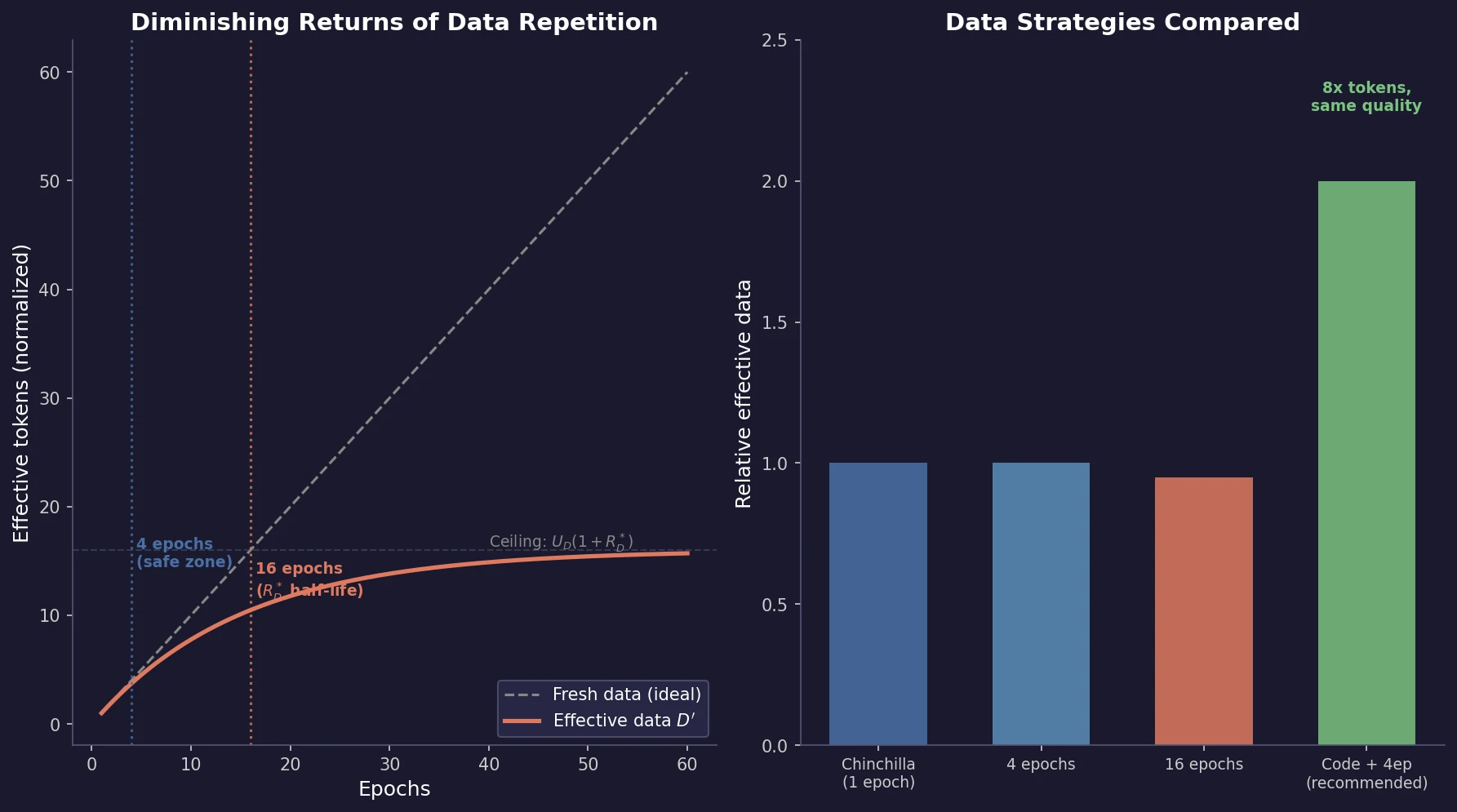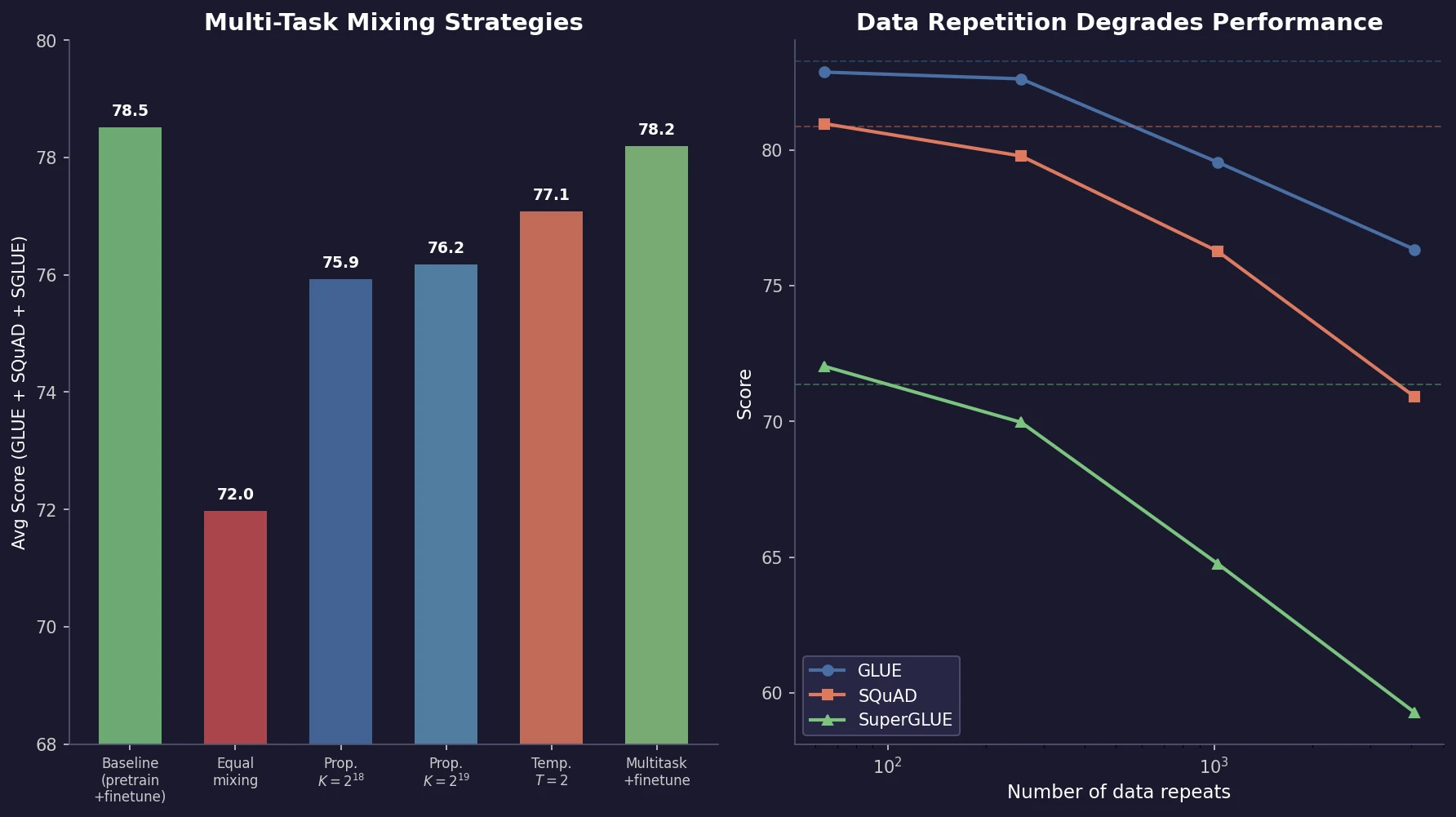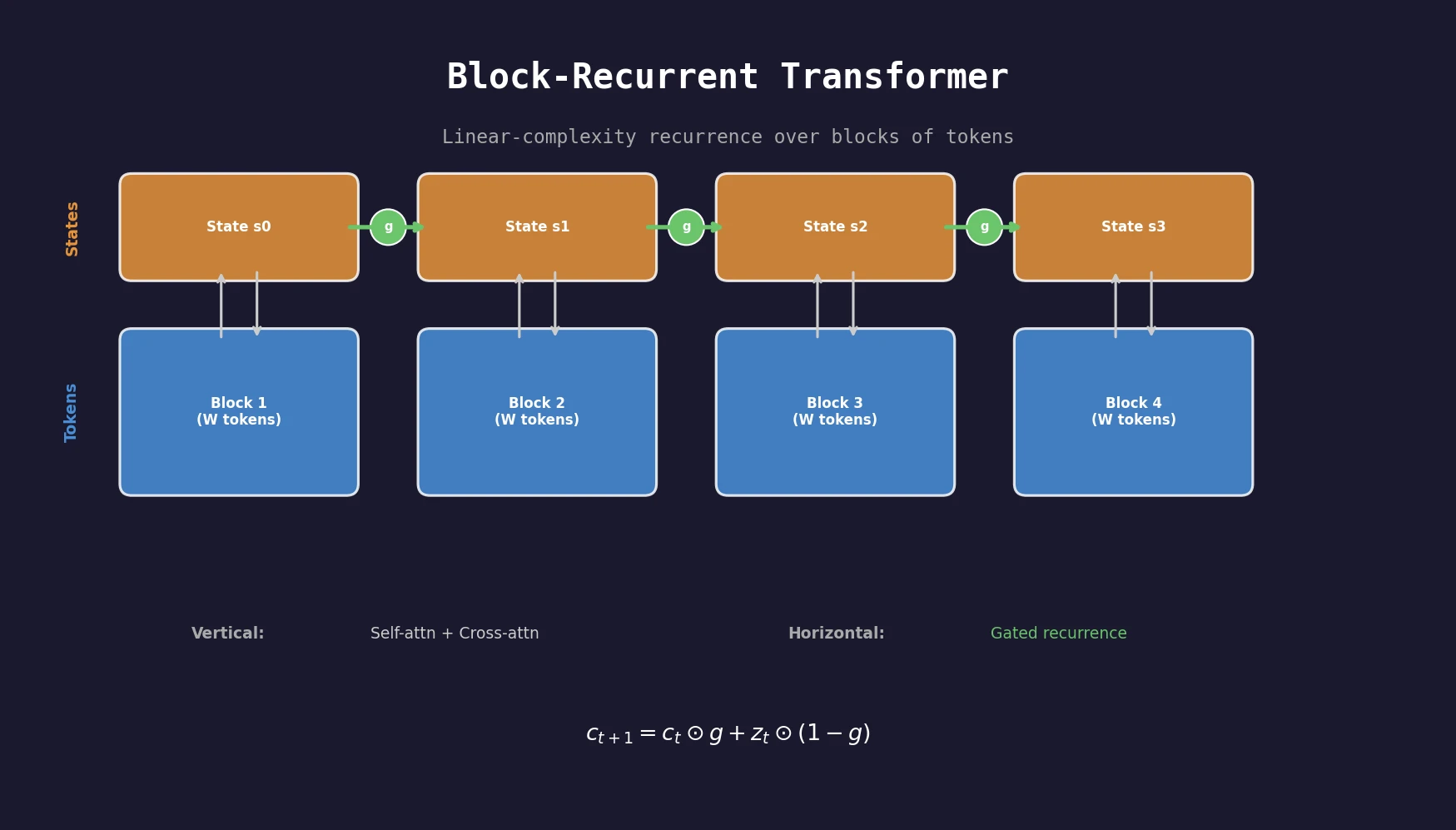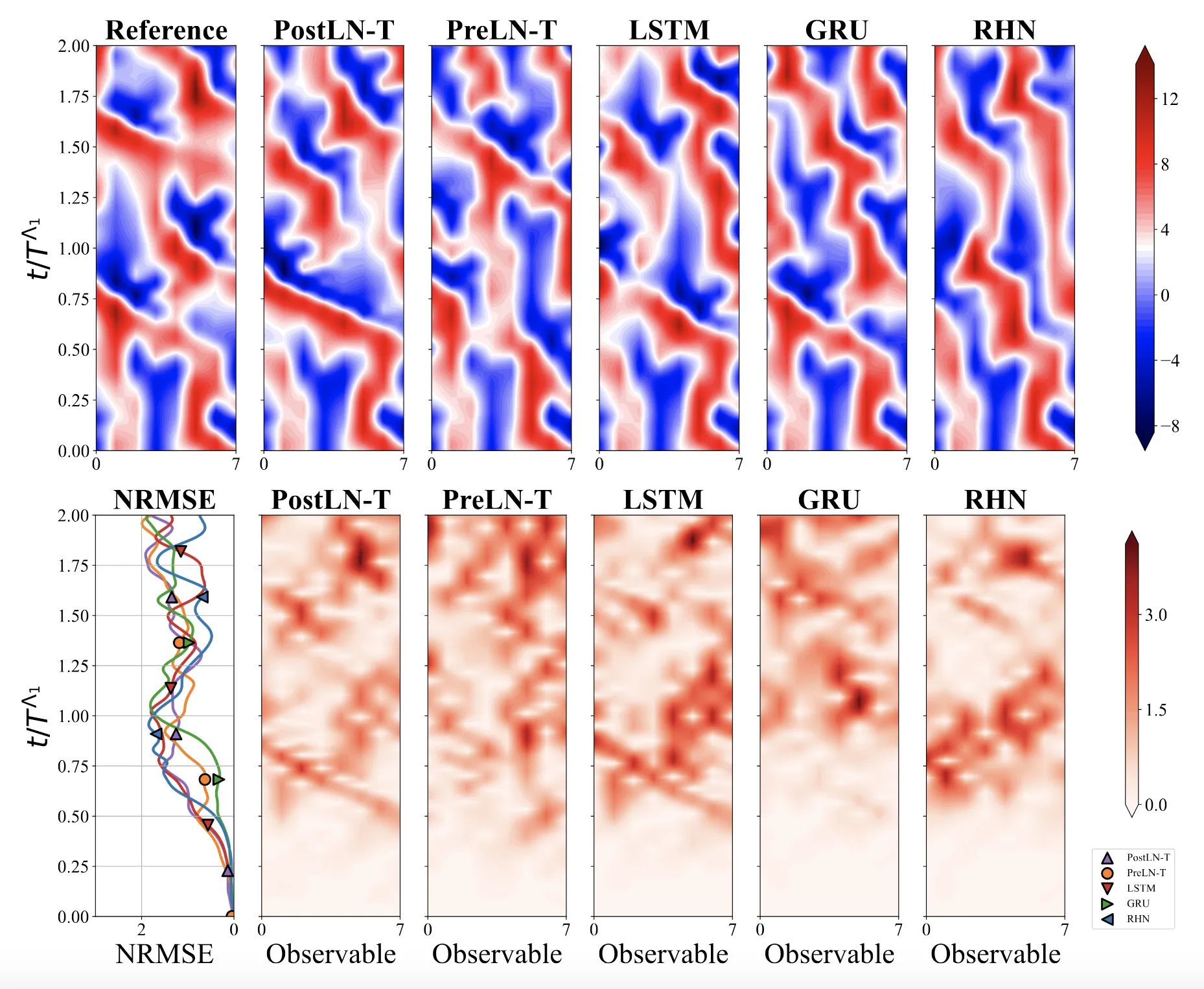
Optimizing Sequence Models for Dynamical Systems
We systematically ablate core mechanisms of Transformers and RNNs, finding that attention-augmented Recurrent Highway Networks outperform standard Transformers on forecasting high-dimensional chaotic systems.