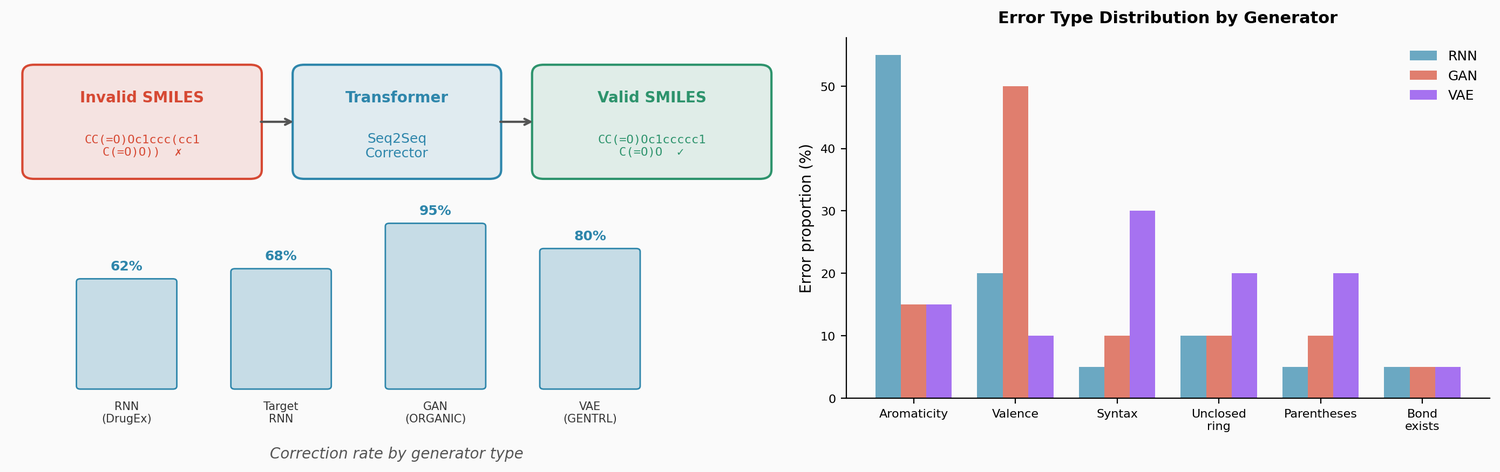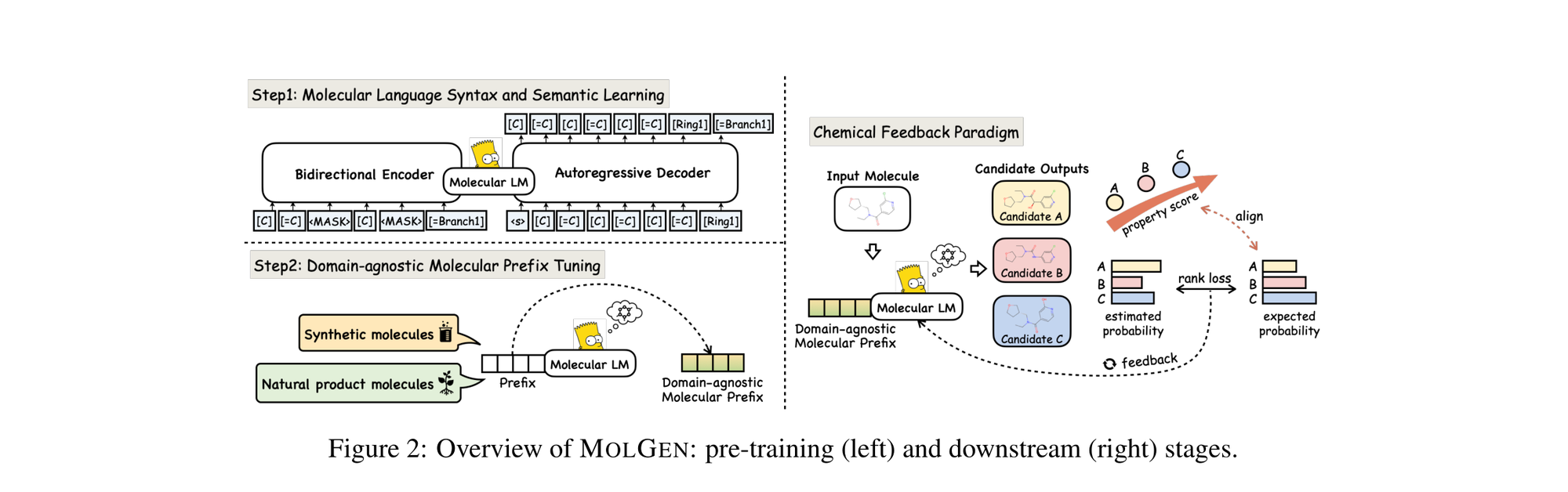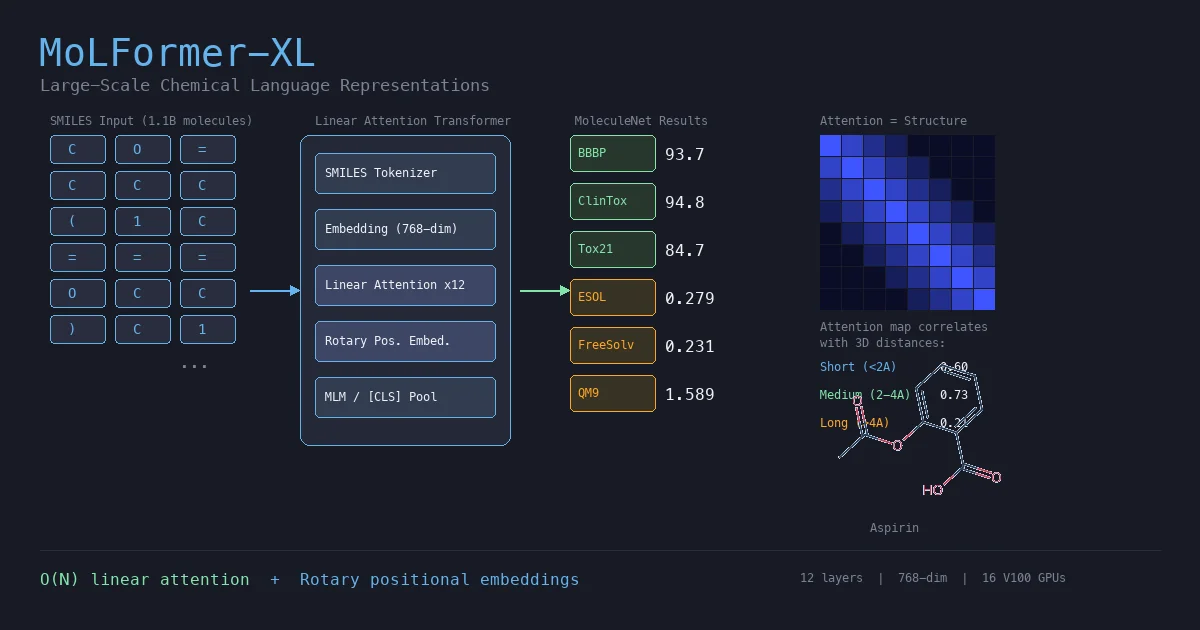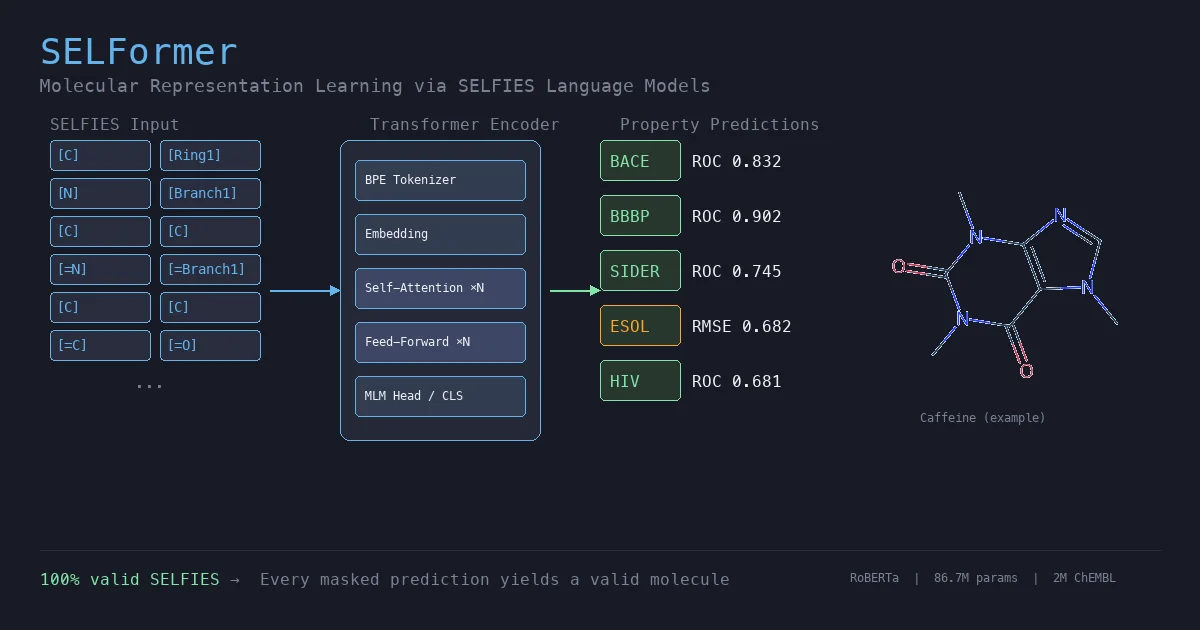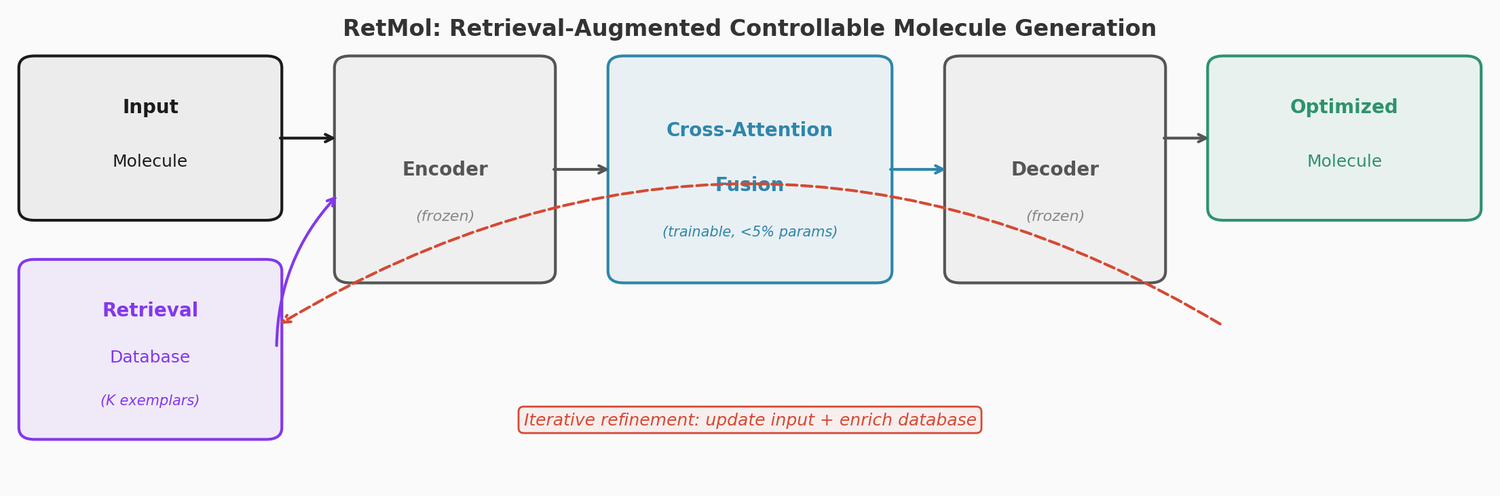
RetMol: Retrieval-Based Controllable Molecule Generation
RetMol plugs a lightweight cross-attention retrieval module into a pre-trained Chemformer backbone to guide molecule generation toward multi-property design criteria. It requires no task-specific fine-tuning and works with as few as 23 exemplar molecules. It achieves 94.5% success on QED optimization, 96.9% on GSK3b/JNK3 dual inhibitor design, and 2.84 kcal/mol average binding affinity improvement on SARS-CoV-2 main protease inhibitor optimization.