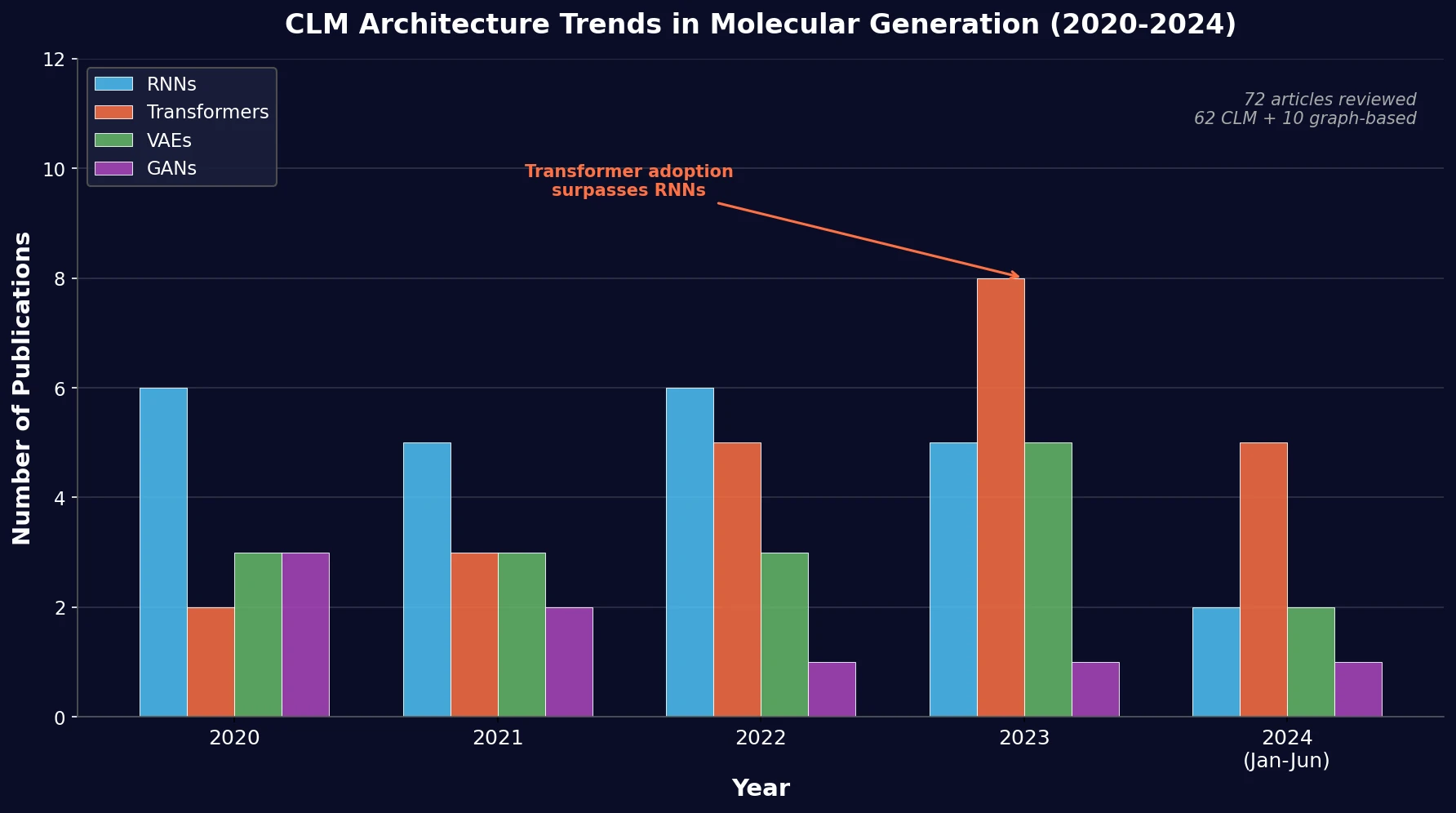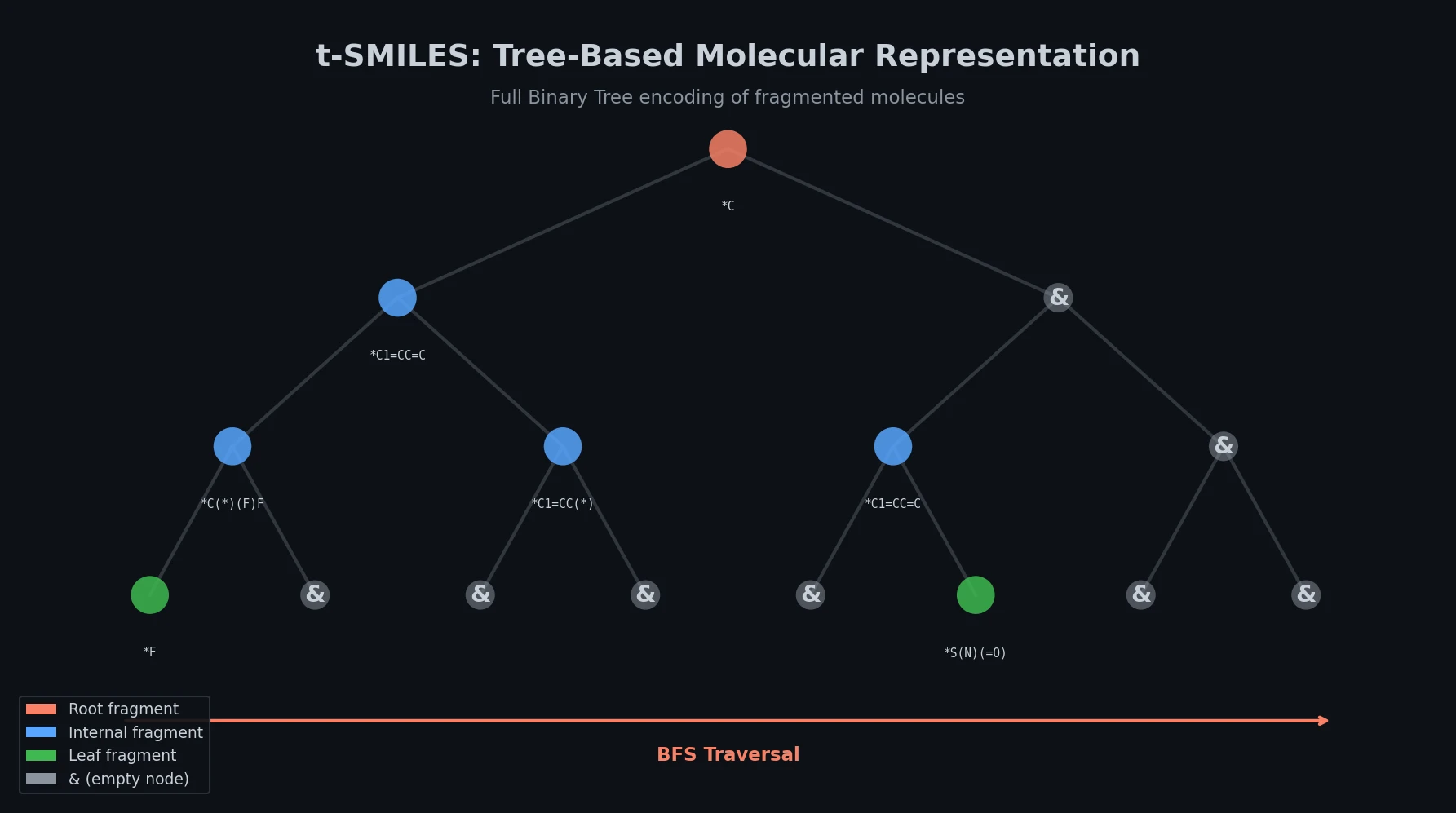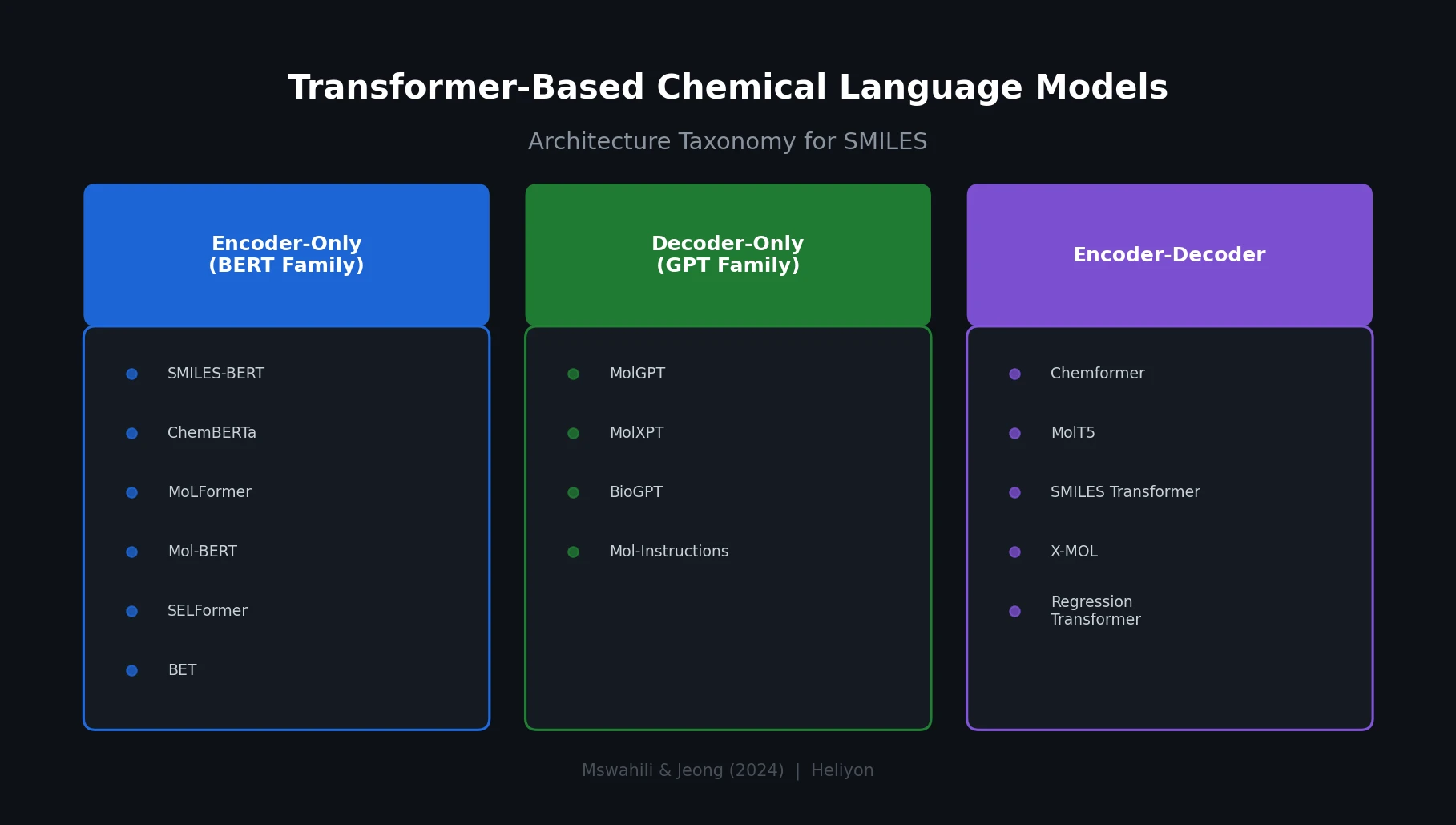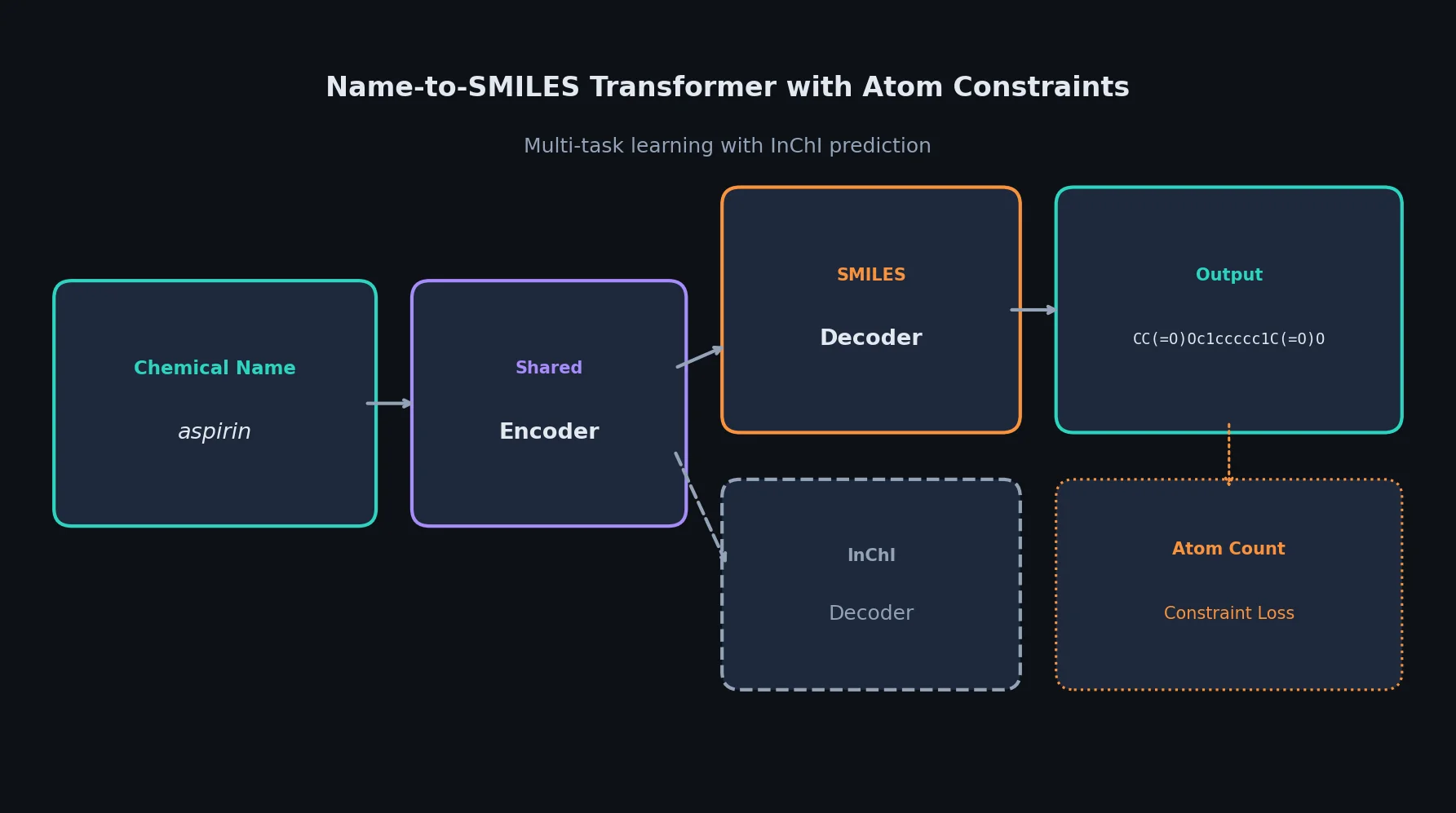
Survey of Scientific LLMs in Bio and Chem Domains
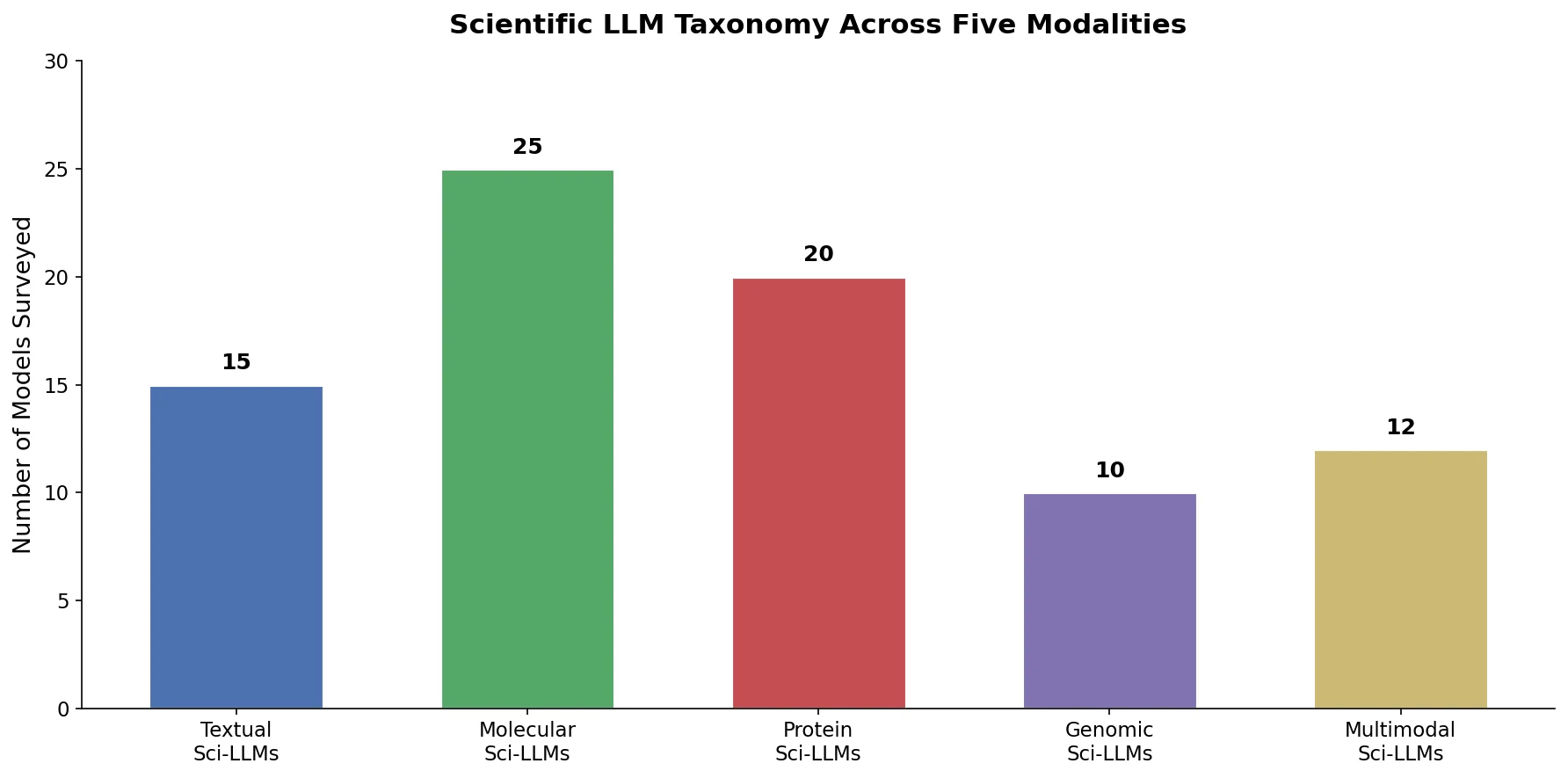
This survey systematically reviews scientific LLMs (Sci-LLMs) across five modalities: textual, molecular, protein, genomic, and multimodal, analyzing architectures, datasets, evaluation methods, and open challenges for AI-driven scientific discovery.