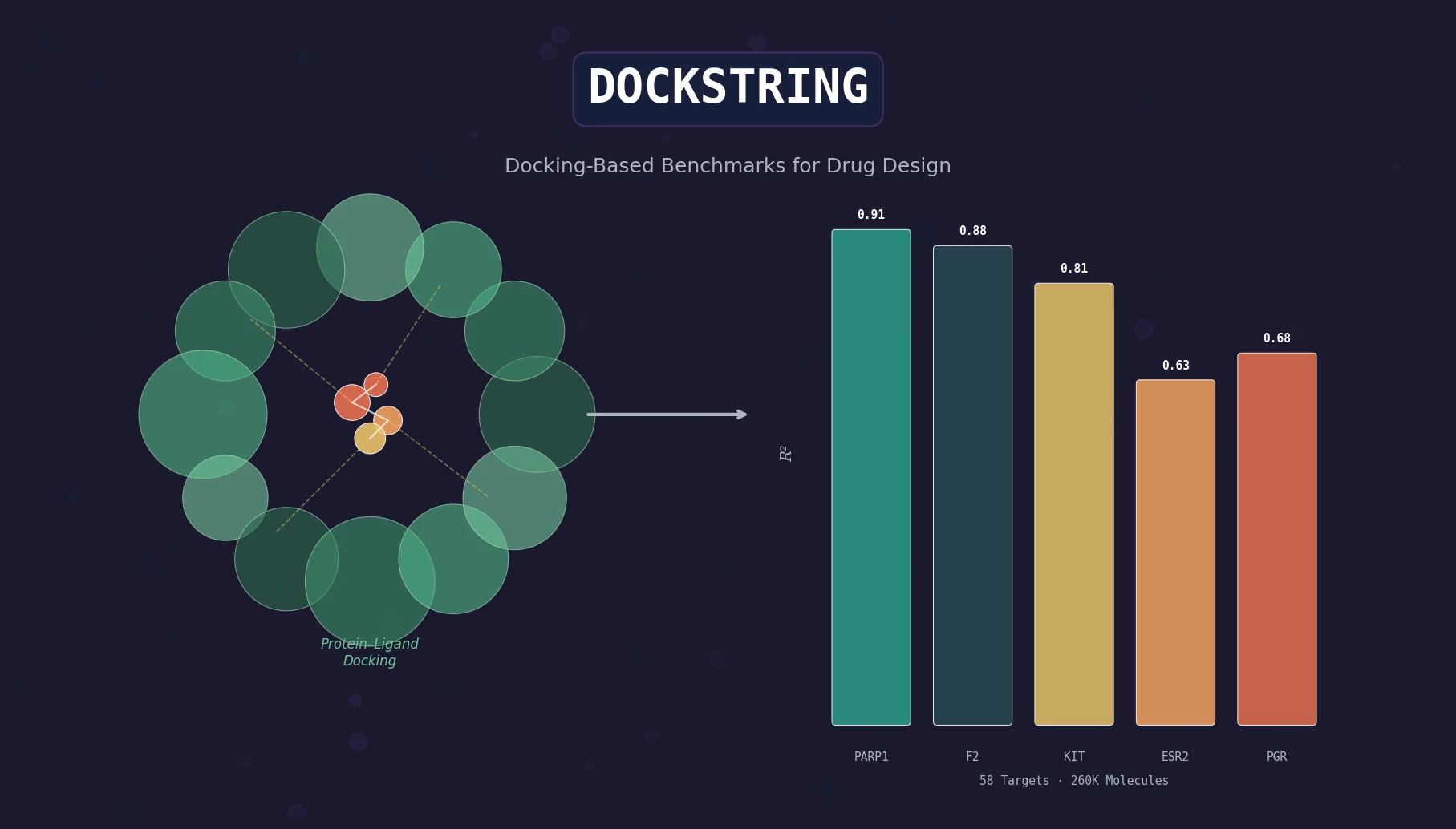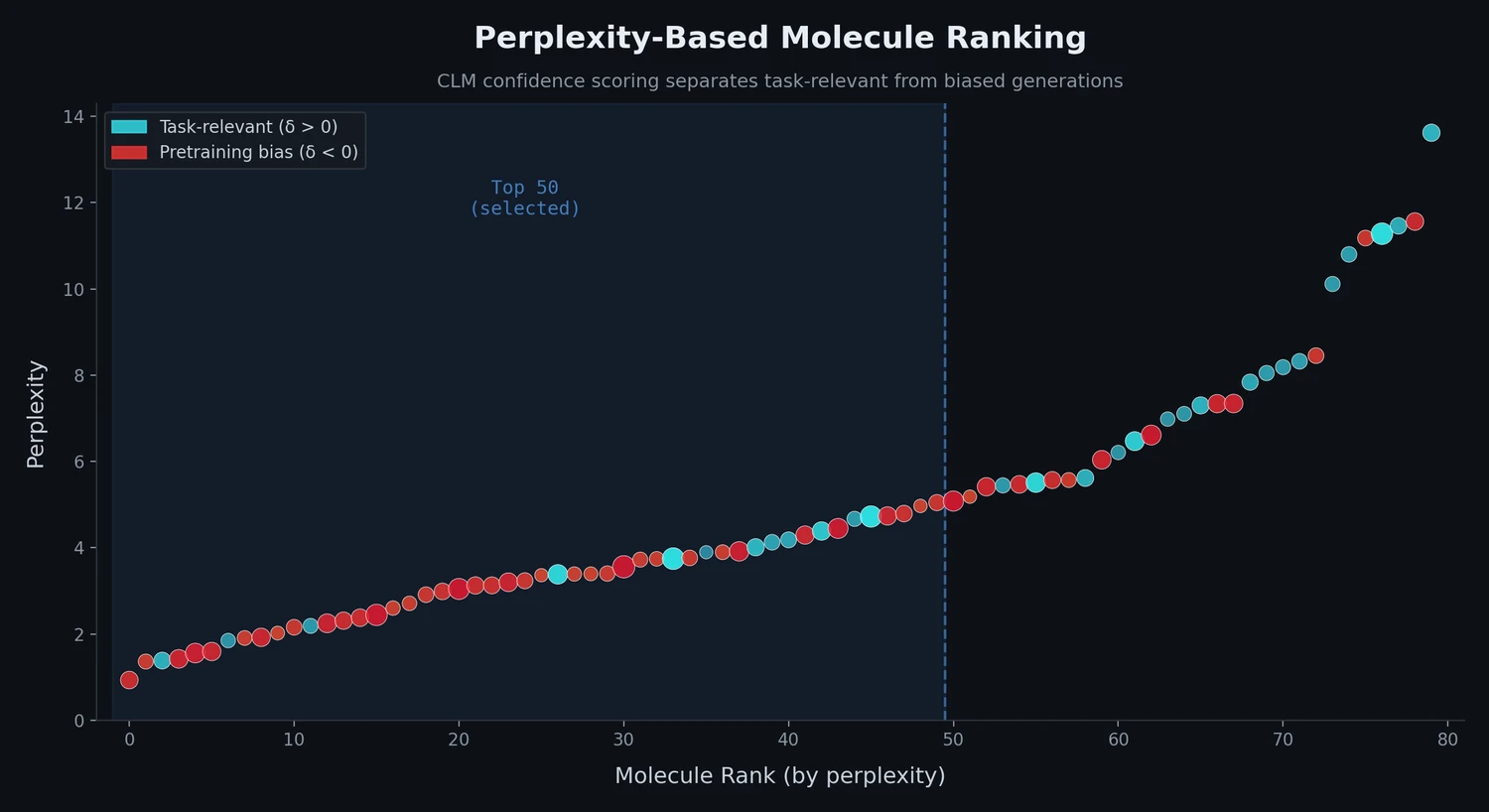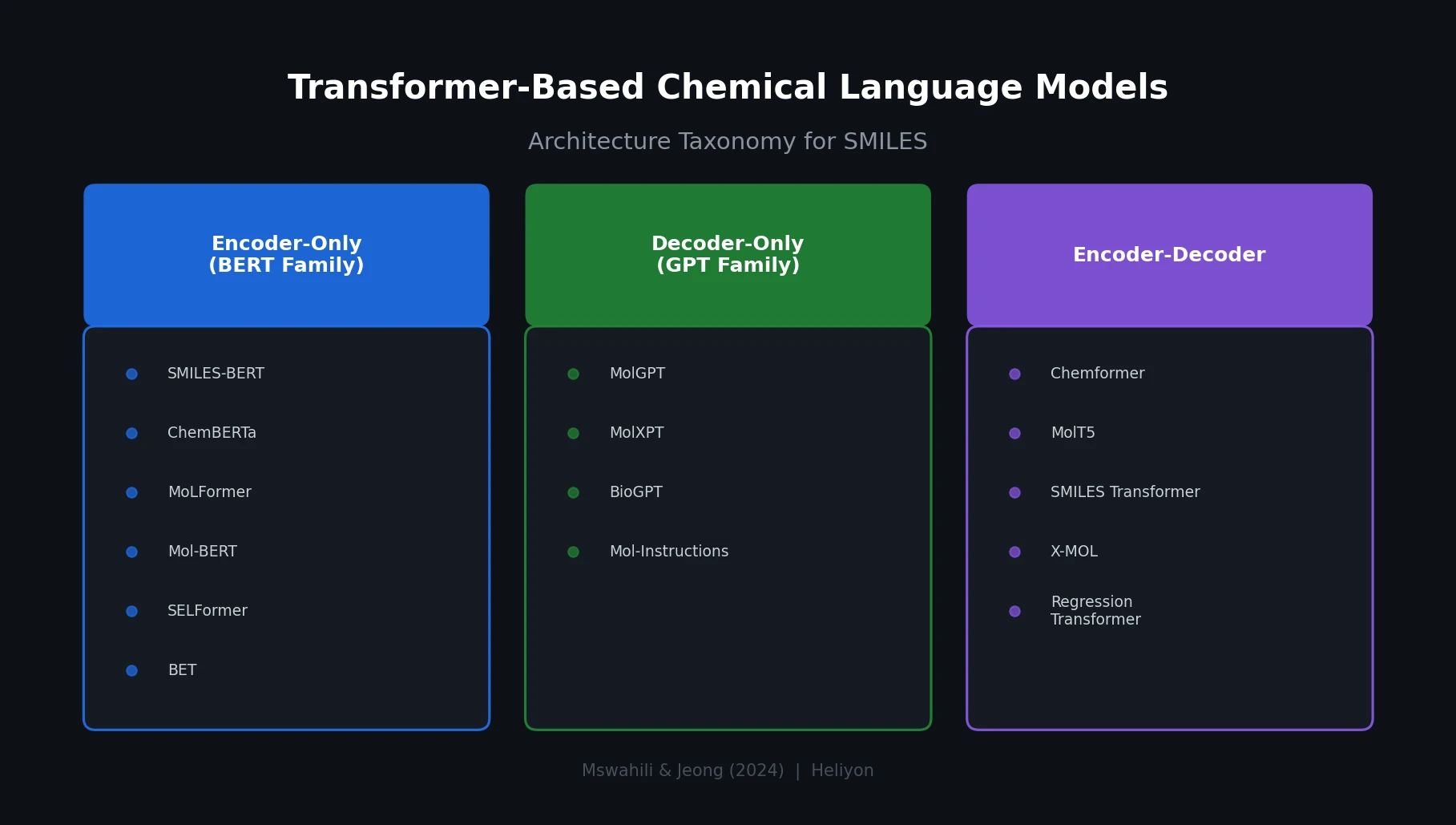
Transformer CLMs for SMILES: Literature Review 2024
A comprehensive review of transformer-based chemical language models operating on SMILES, categorizing encoder-only (BERT variants), decoder-only (GPT variants), and encoder-decoder models with analysis of tokenization strategies, pre-training approaches, and future directions.