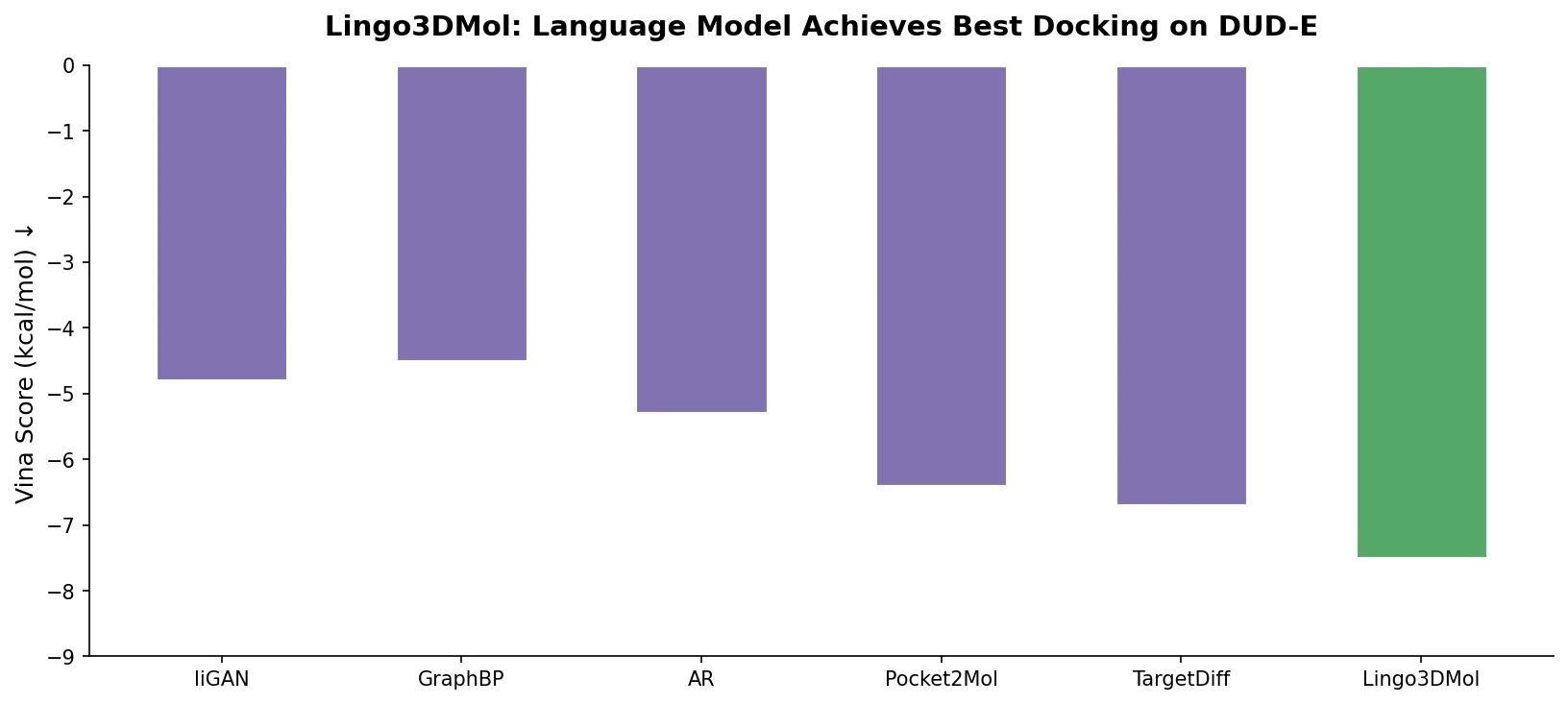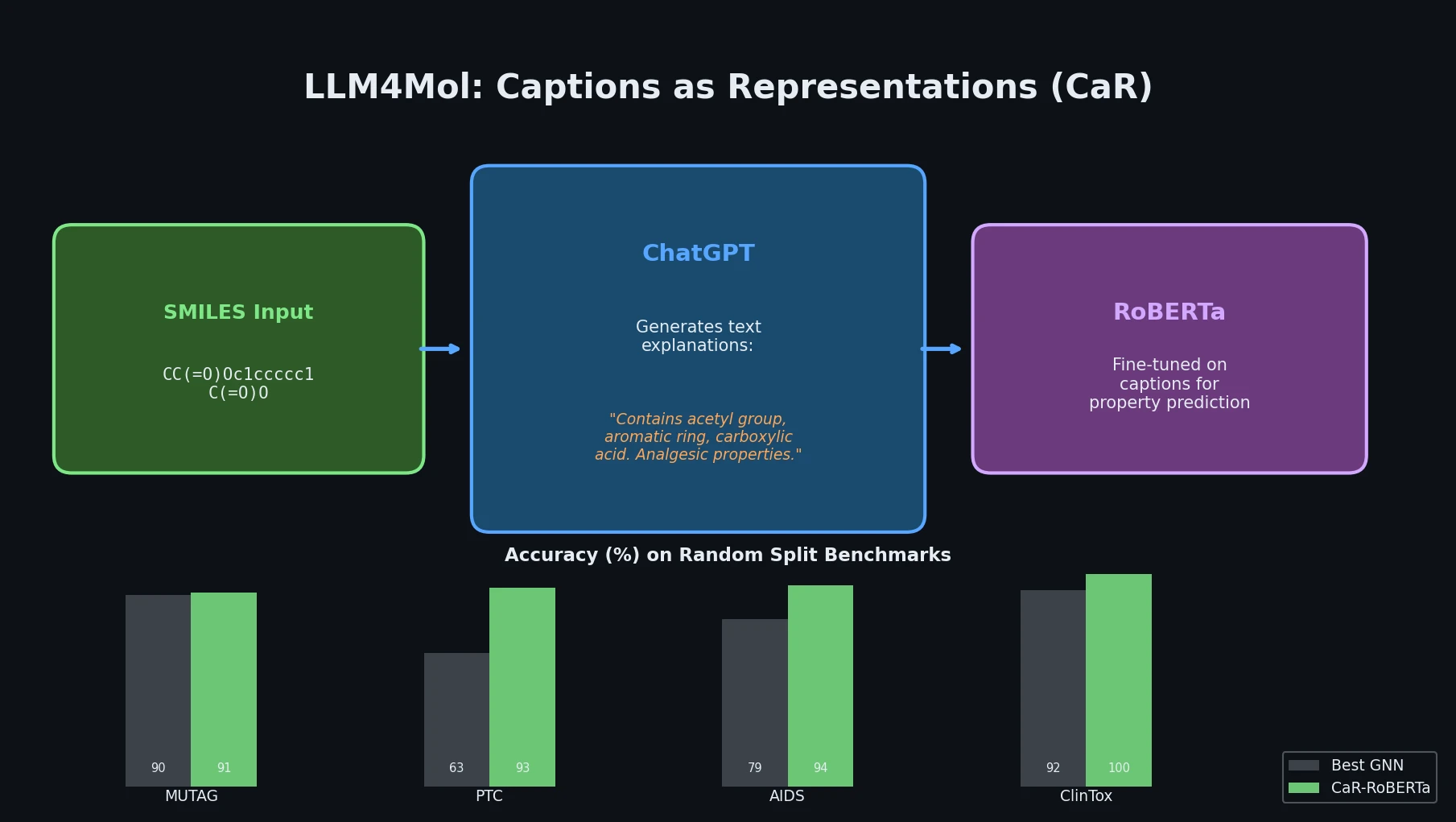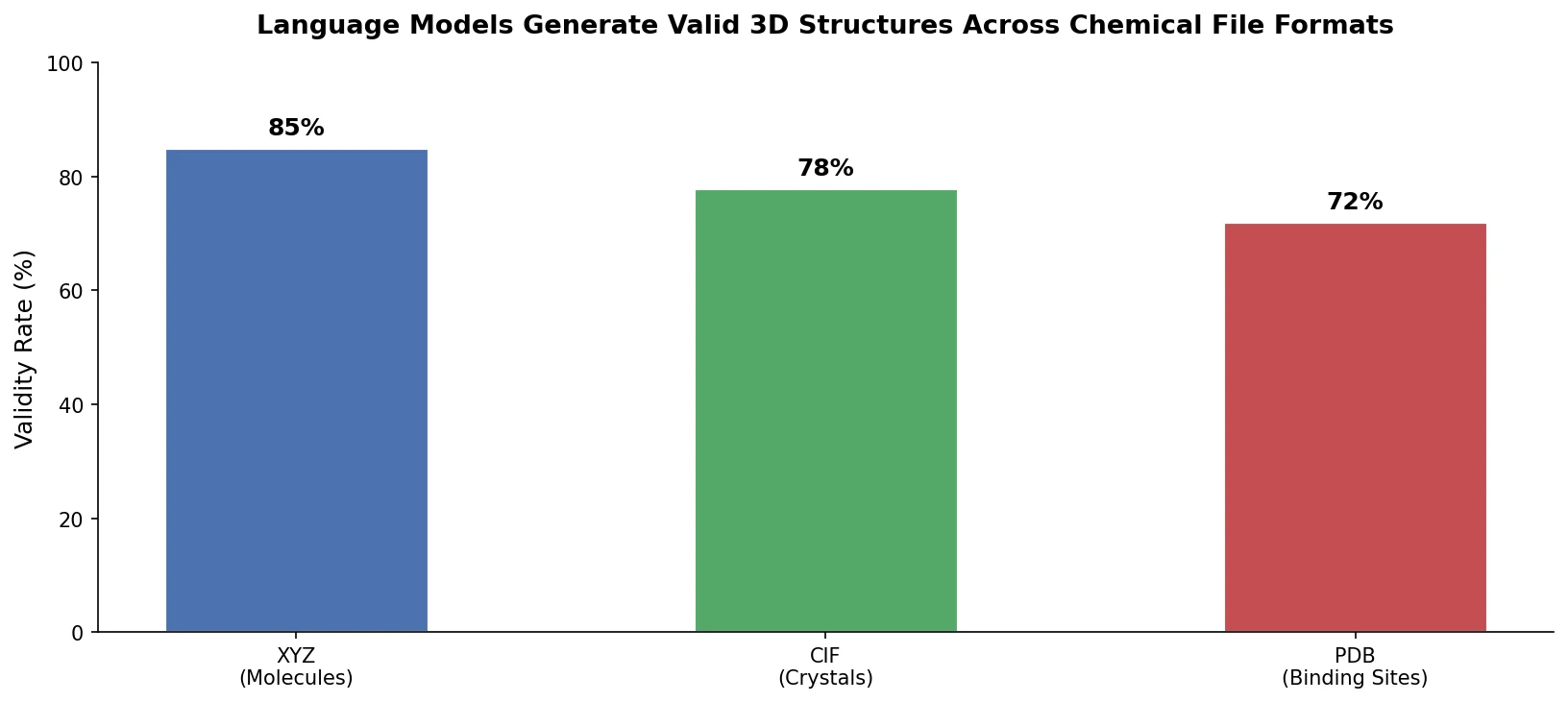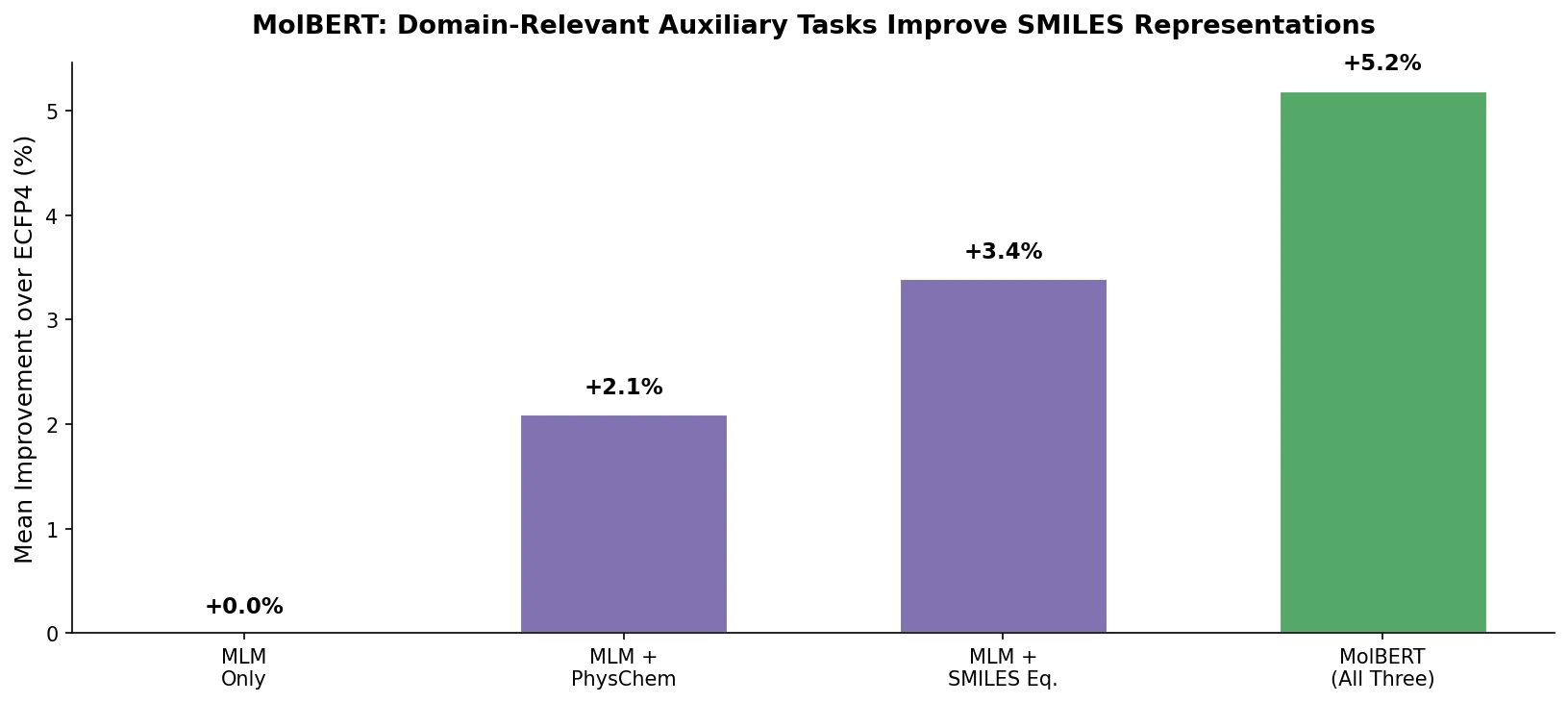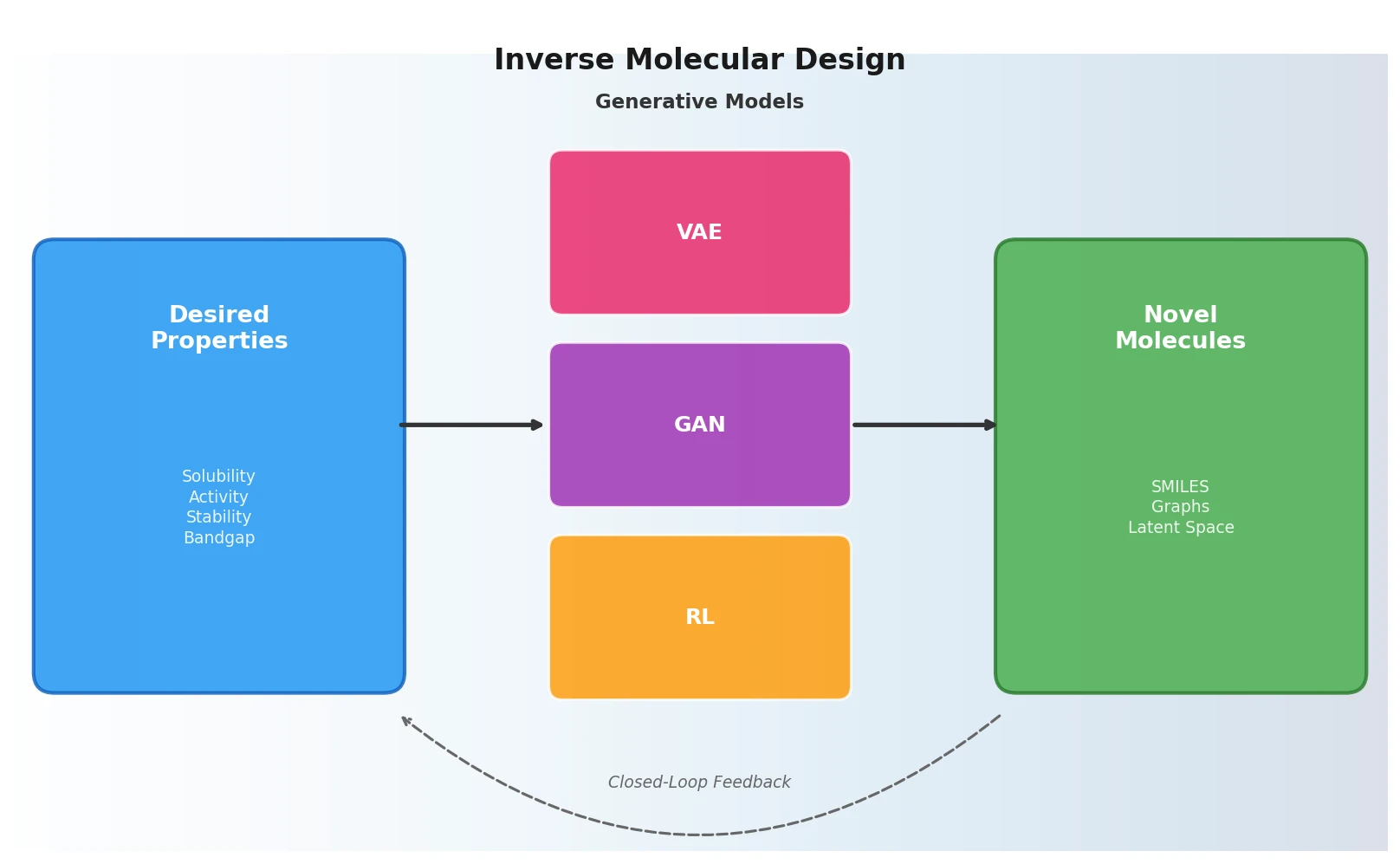
Inverse Molecular Design with ML Generative Models
A foundational review surveying how deep generative models (VAEs, GANs, reinforcement learning) enable inverse molecular design, covering molecular representations, chemical space navigation, and applications from drug discovery to materials engineering.