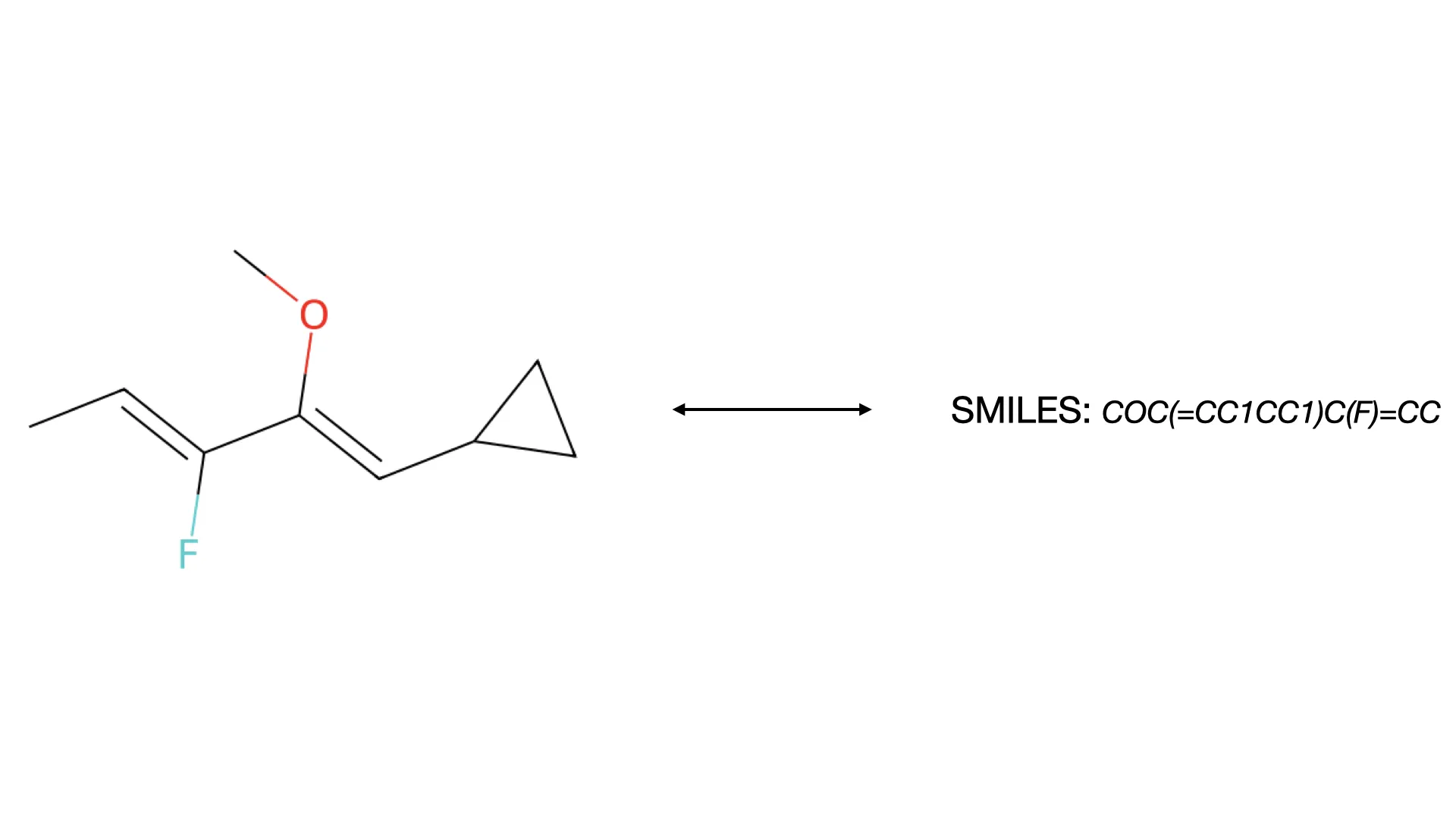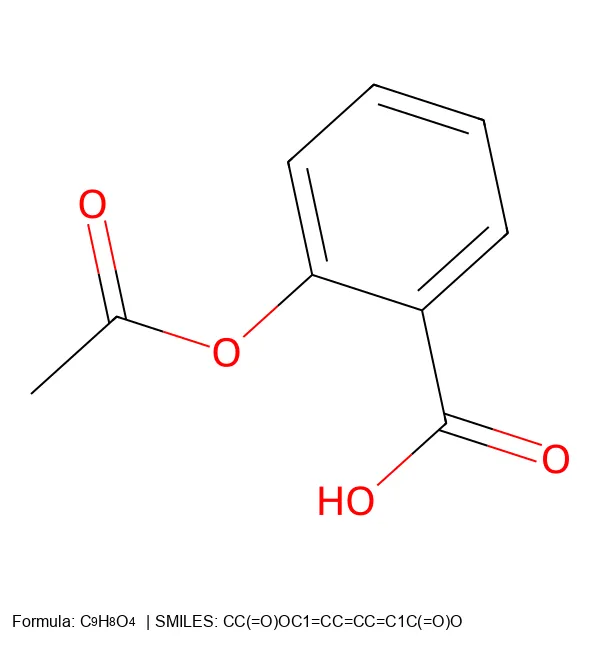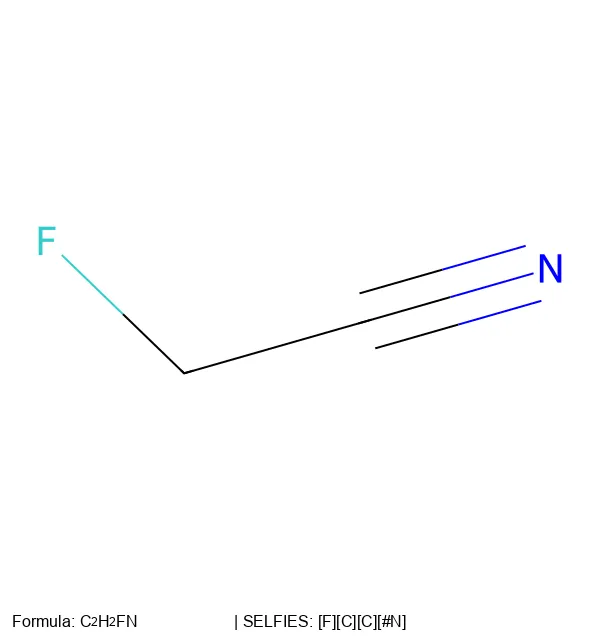
MolParser-7M & WildMol: Large-Scale OCSR Datasets
The MolParser project introduces two key datasets: MolParser-7M, the largest training dataset for Optical Chemical Structure Recognition (OCSR) with 7.7M pairs of images and E-SMILES strings, and WildMol, a new 20k-sample benchmark for evaluating models on challenging real-world data. The training data uniquely combines millions of diverse synthetic molecules with 400,000 manually annotated in-the-wild samples.