Copper Adatom Diffusion on Cu(100): LAMMPS Simulation
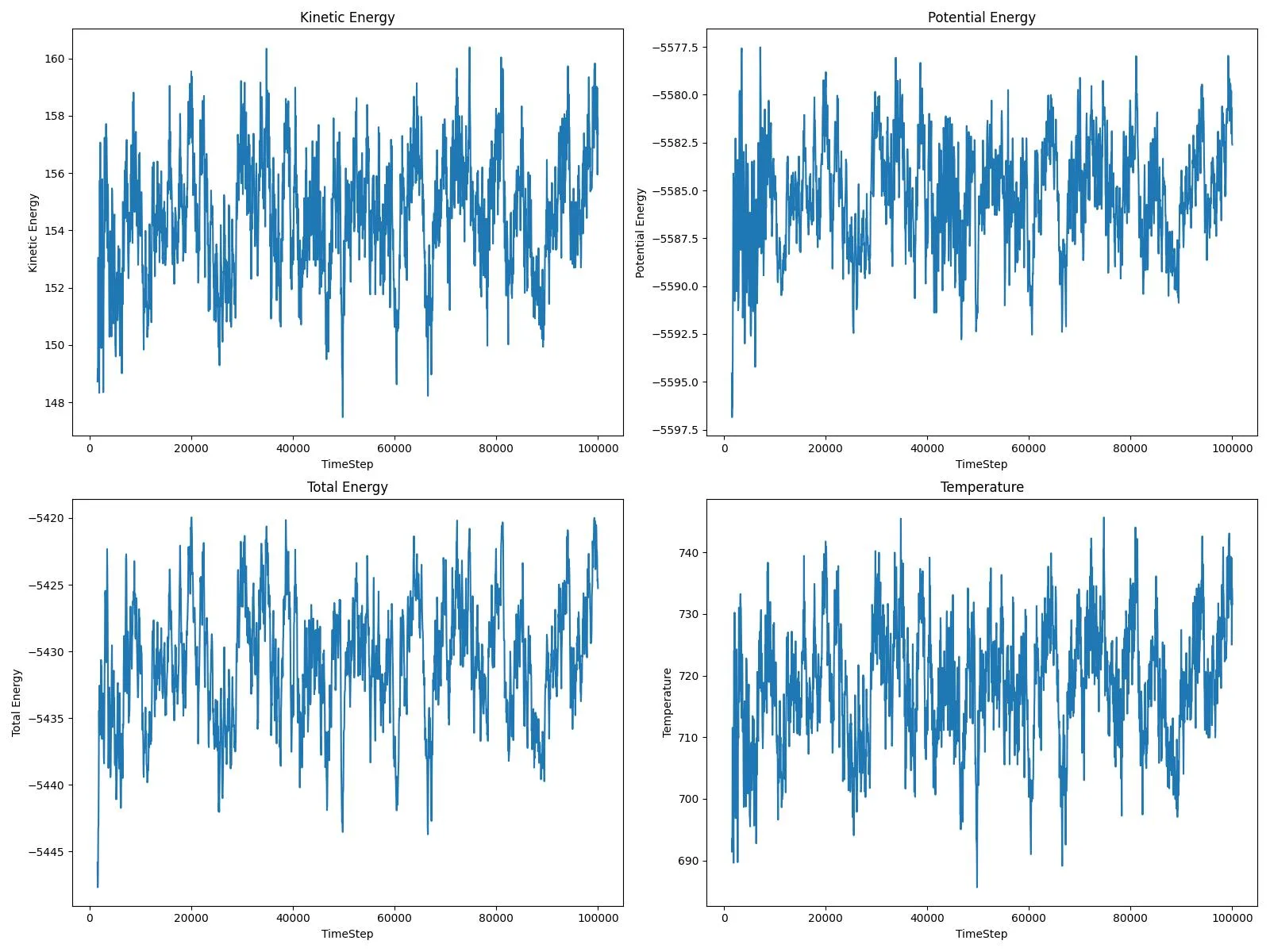
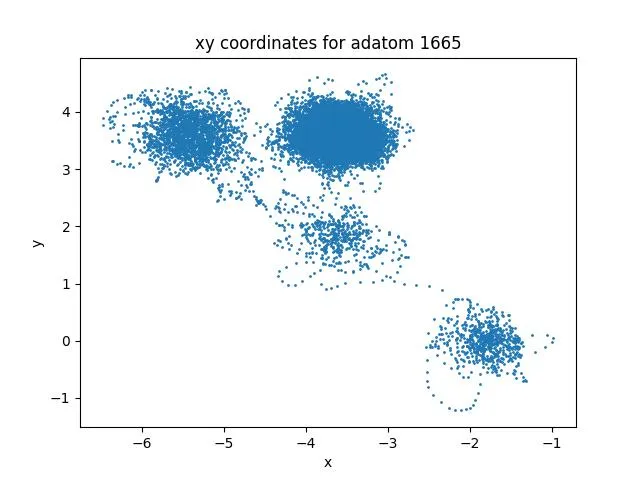
Watch copper atoms move across a crystal surface in this molecular dynamics simulation. This video demonstrates surface diffusion mechanisms important for understanding catalysis and crystal growth processes.