Correlations in the Motion of Atoms in Liquid Argon
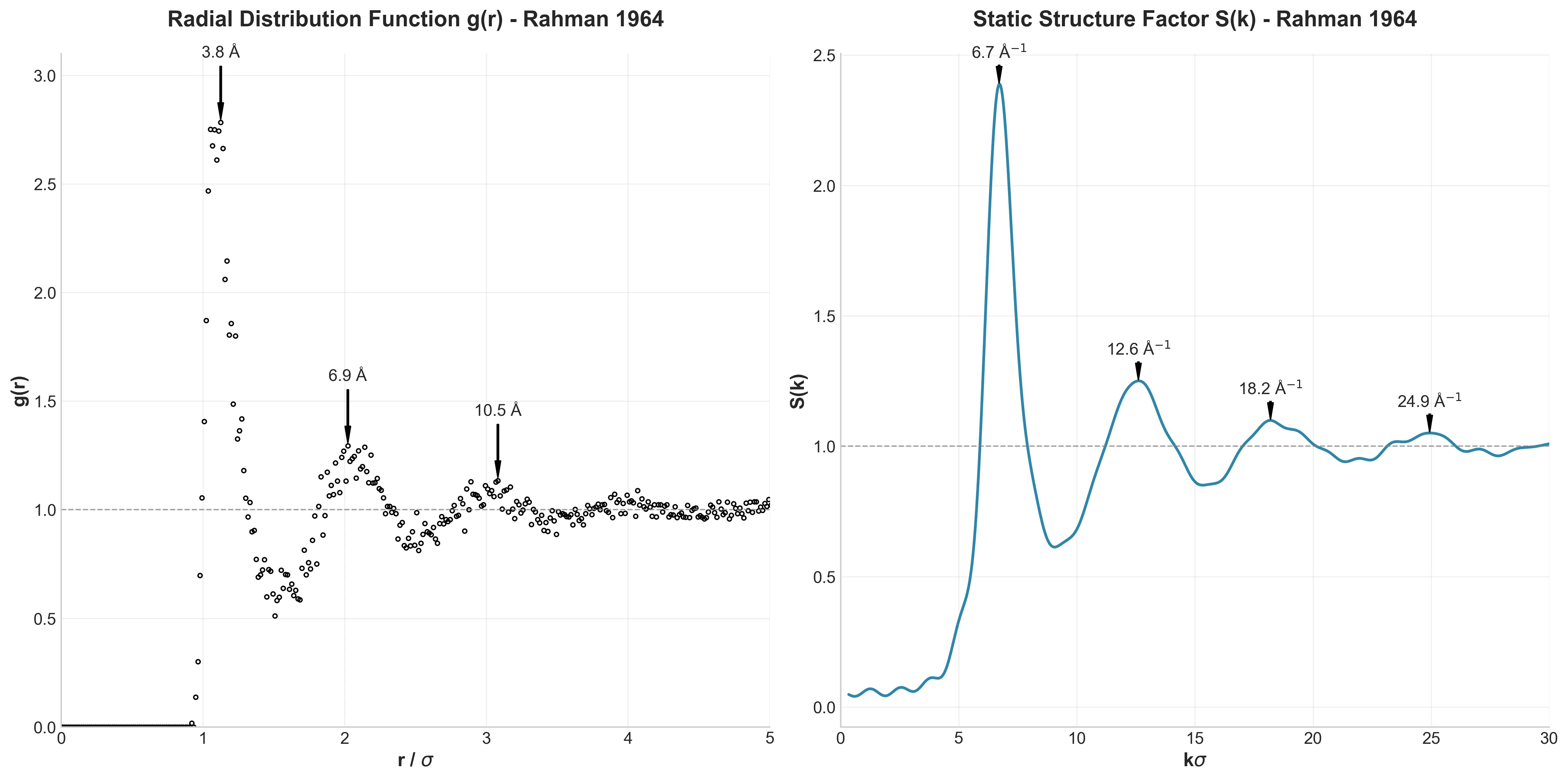
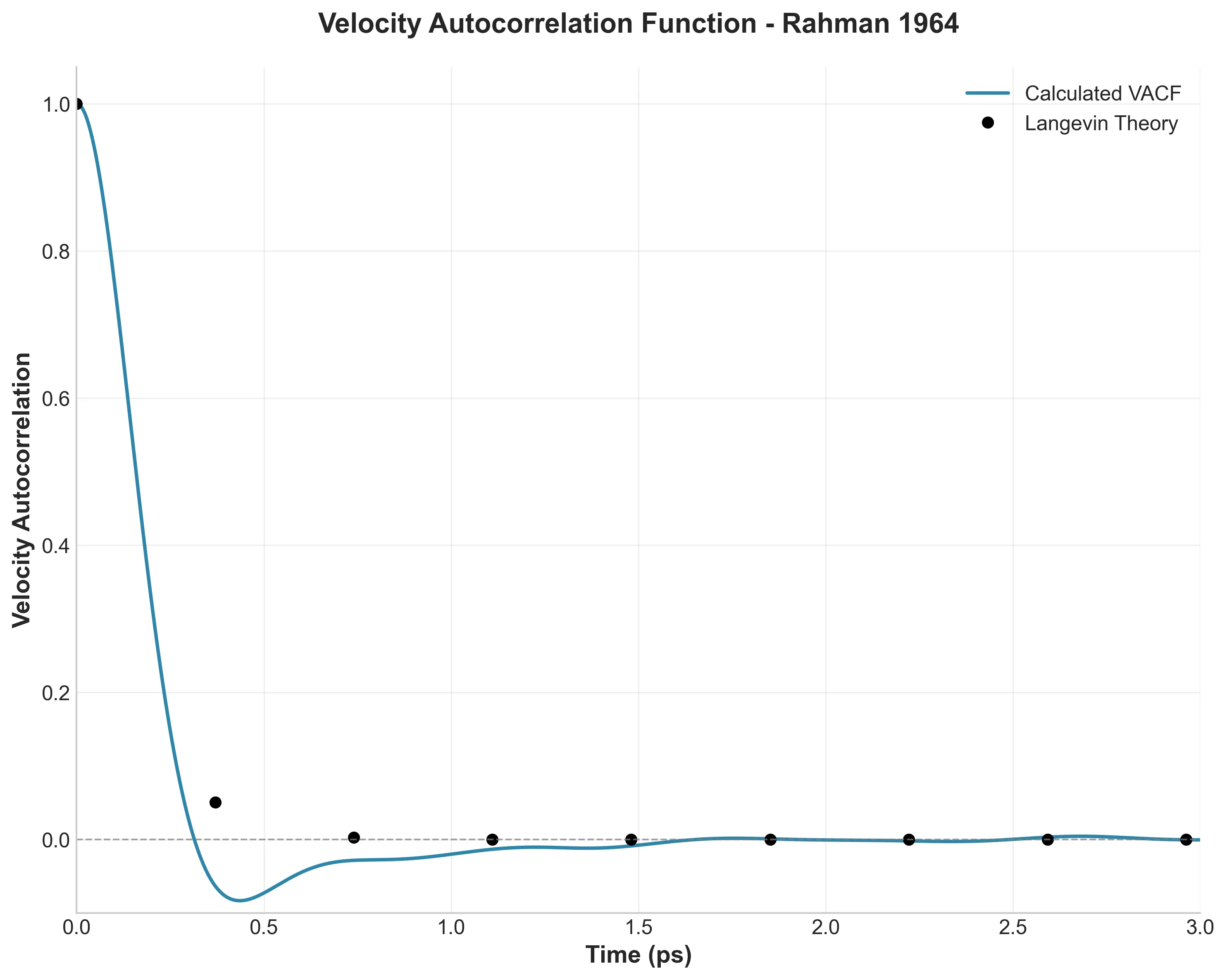
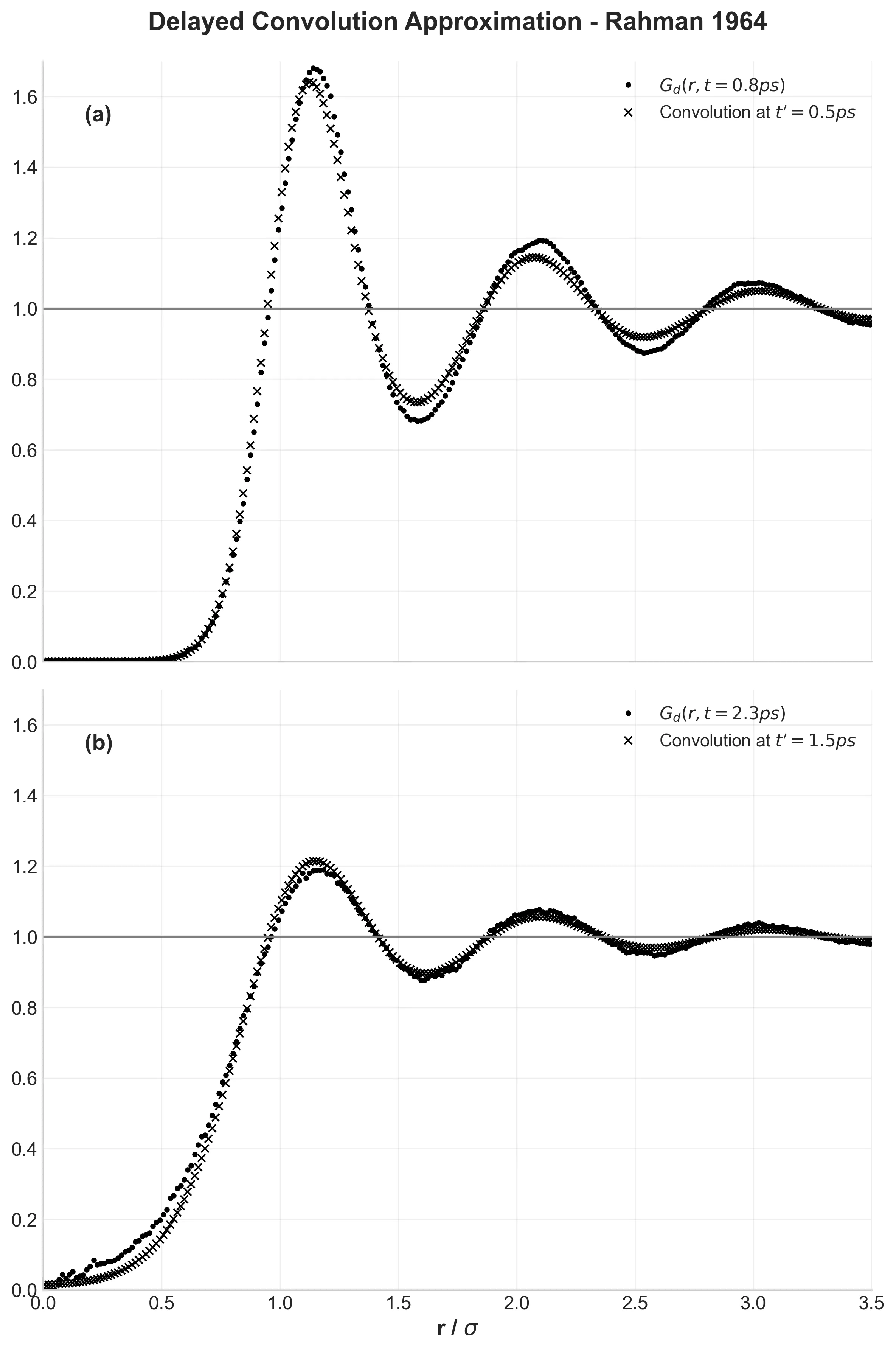
This work validated classical Molecular Dynamics for simulating liquids, revealing the ‘cage effect’ in velocity autocorrelation and establishing predictor-corrector integration algorithms for N-body problems.