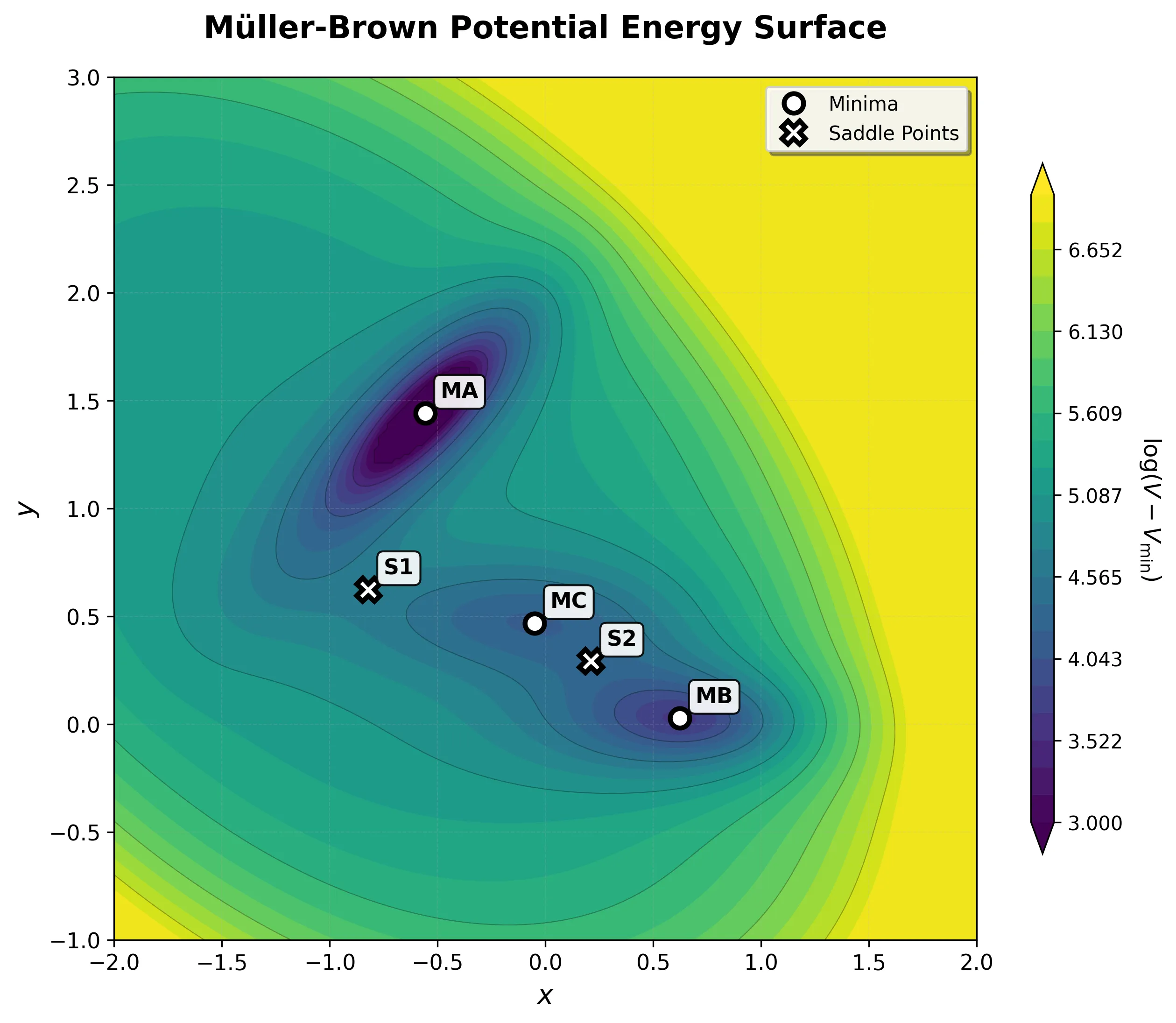
Müller-Brown Basin MB: Langevin Dynamics Simulation
Watch particle dynamics in the product minimum of the Müller-Brown potential. This simulation shows intermediate thermal motion behavior at -108.17 kJ/mol energy level.
Watch particle dynamics in the product minimum of the Müller-Brown potential. This simulation shows intermediate thermal motion behavior at -108.17 kJ/mol energy level.
A high-performance, GPU-accelerated PyTorch testbed for ML-MD algorithms featuring JIT-compiled analytical Jacobian force kernels achieving 3-10x speedup over autograd, robust Langevin dynamics with Velocity-Verlet integration, and modular architecture designed as ground-truth validation for novel machine learning approaches in molecular dynamics.
Experience rare transition events between energy basins in this extended Müller-Brown simulation. Watch as particles overcome energy barriers to explore different regions of the potential energy landscape.
ICLR 2025 paper introducing DenoiseVAE, which learns adaptive, atom-specific noise distributions through a VAE framework to improve denoising-based pre-training for molecular force field prediction, outperforming fixed Gaussian noise approaches on quantum chemistry benchmarks.
ICML 2025 analysis rigorously quantifying when non-conservative force models (which predict forces directly) fail in molecular dynamics, demonstrating simulation instabilities and proposing hybrid architectures that capture speed benefits without sacrificing physical correctness.
ICML 2025 paper proposing energy conservation metrics as critical diagnostics for machine learning interatomic potentials and introducing eSEN, a novel architecture designed to bridge the gap between test-set accuracy and real simulation performance on materials property prediction.
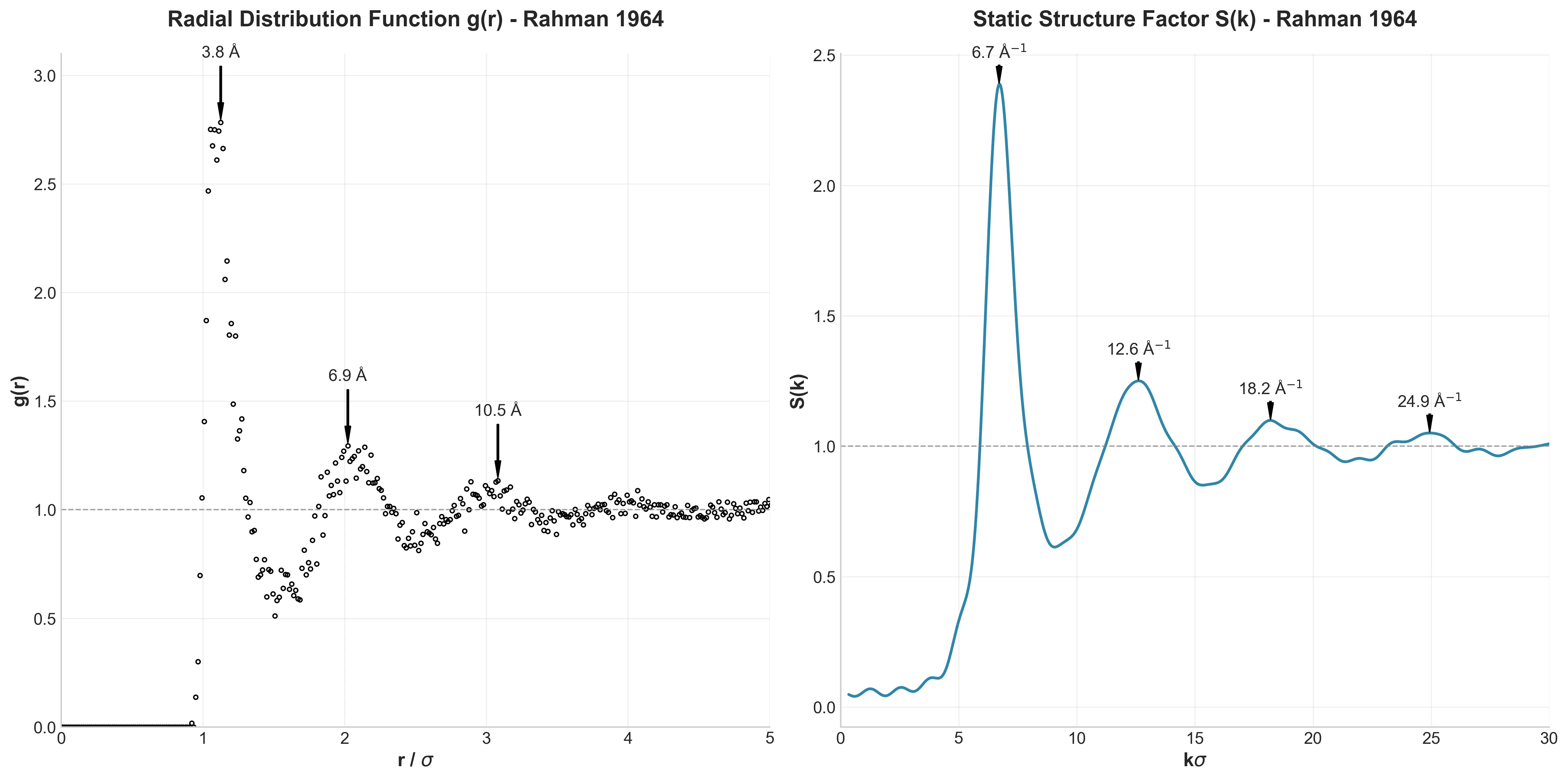
Explore the molecular dynamics of liquid argon in this fundamental LAMMPS simulation. This classic system demonstrates liquid-state behavior and serves as a benchmark for molecular dynamics methods.
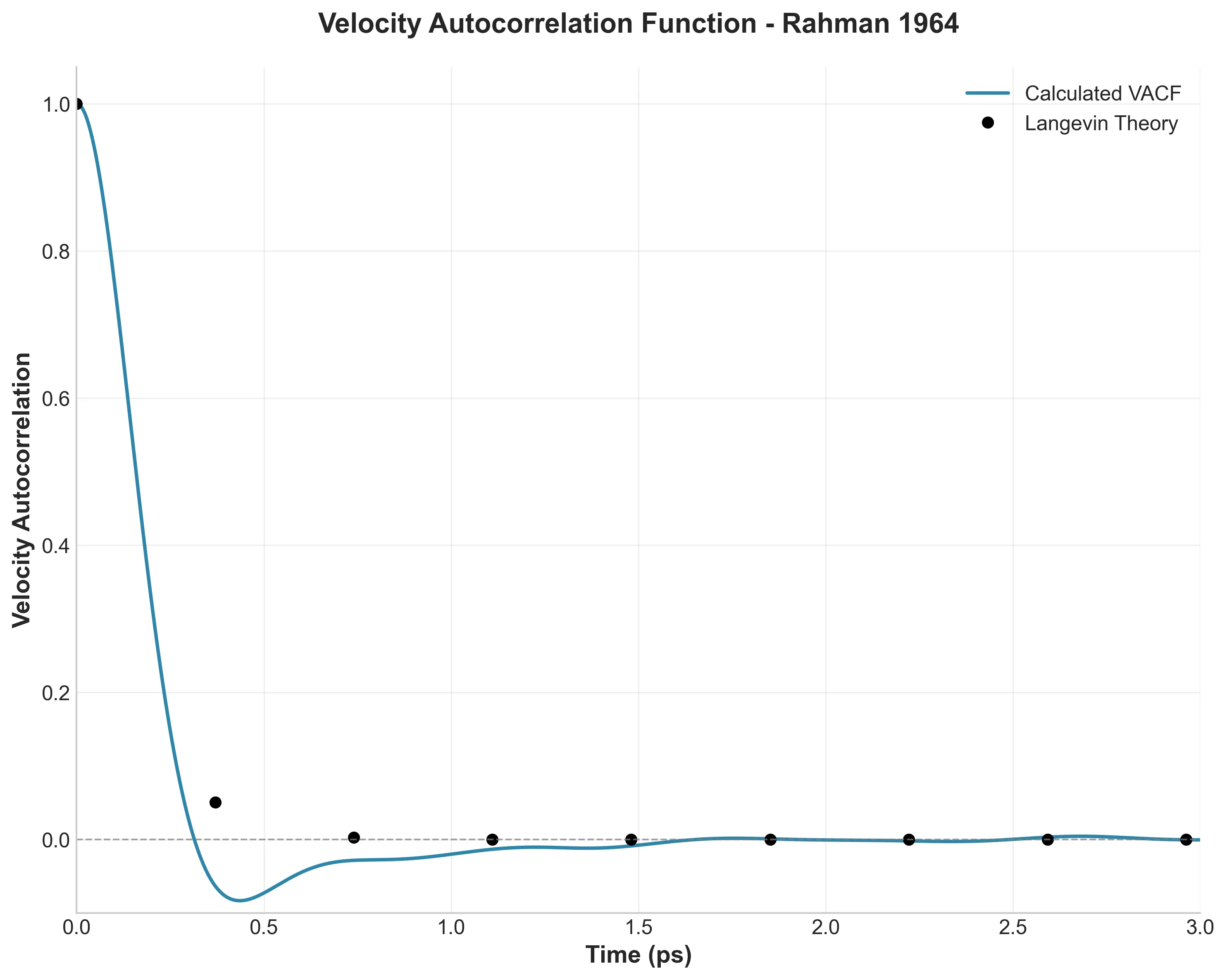
A digital restoration of Rahman’s seminal 1964 molecular dynamics paper using LAMMPS and a production-grade Python analysis pipeline featuring intelligent decorator-based caching, fully vectorized NumPy computations for O(N^2) operations, and modern tooling (uv, type hints, Makefile automation) transforming academic scripts into reproducible research toolkit.
I replicated Rahman’s landmark 1964 liquid argon molecular dynamics simulation using modern tools, building a production-grade Python analysis pipeline with intelligent caching, vectorization, and type safety to bridge vintage science with contemporary software engineering.
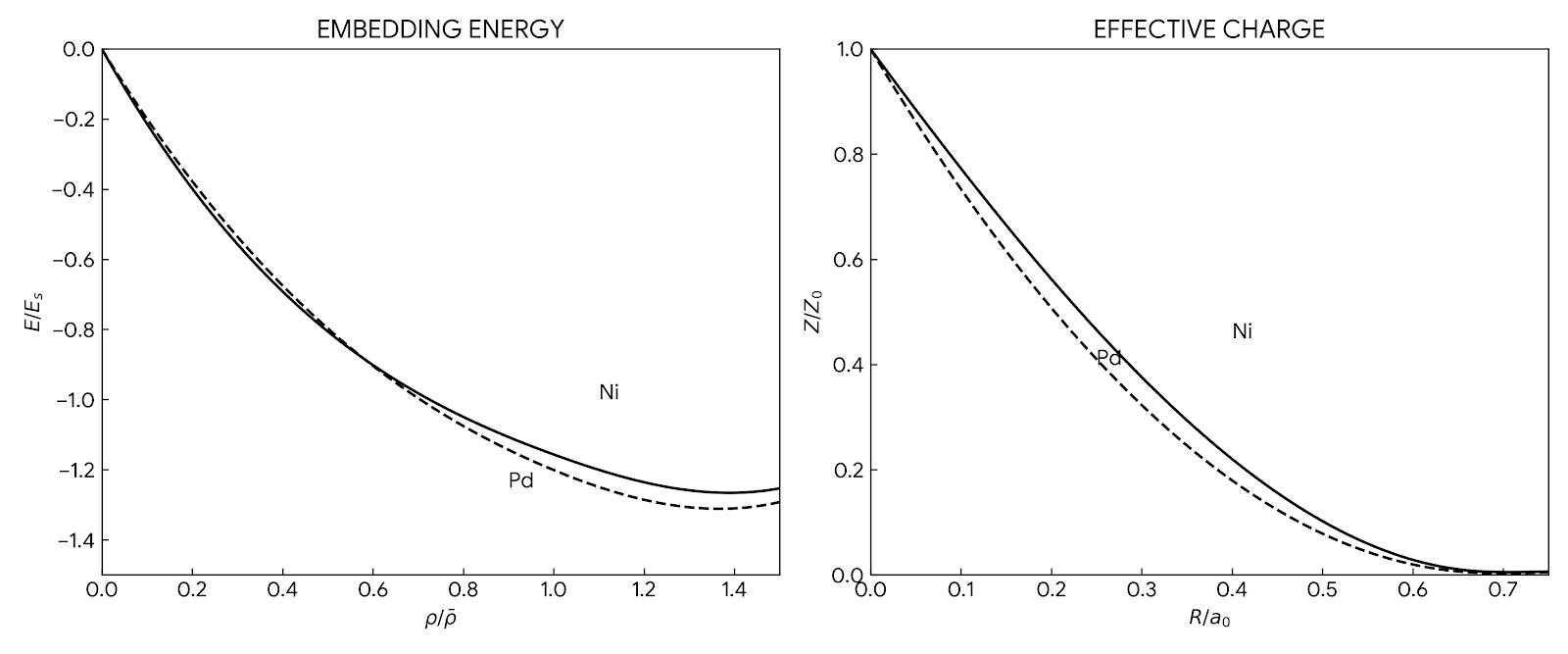
The foundational 1984 paper introducing EAM, a semi-empirical many-body interatomic potential that incorporates density functional theory concepts to accurately simulate metallic systems while maintaining computational efficiency comparable to pair potentials.
Torrie and Valleau’s 1977 paper introducing importance sampling with non-physical distributions to overcome the sampling gap problem in Monte Carlo free-energy calculations, particularly for phase transitions.
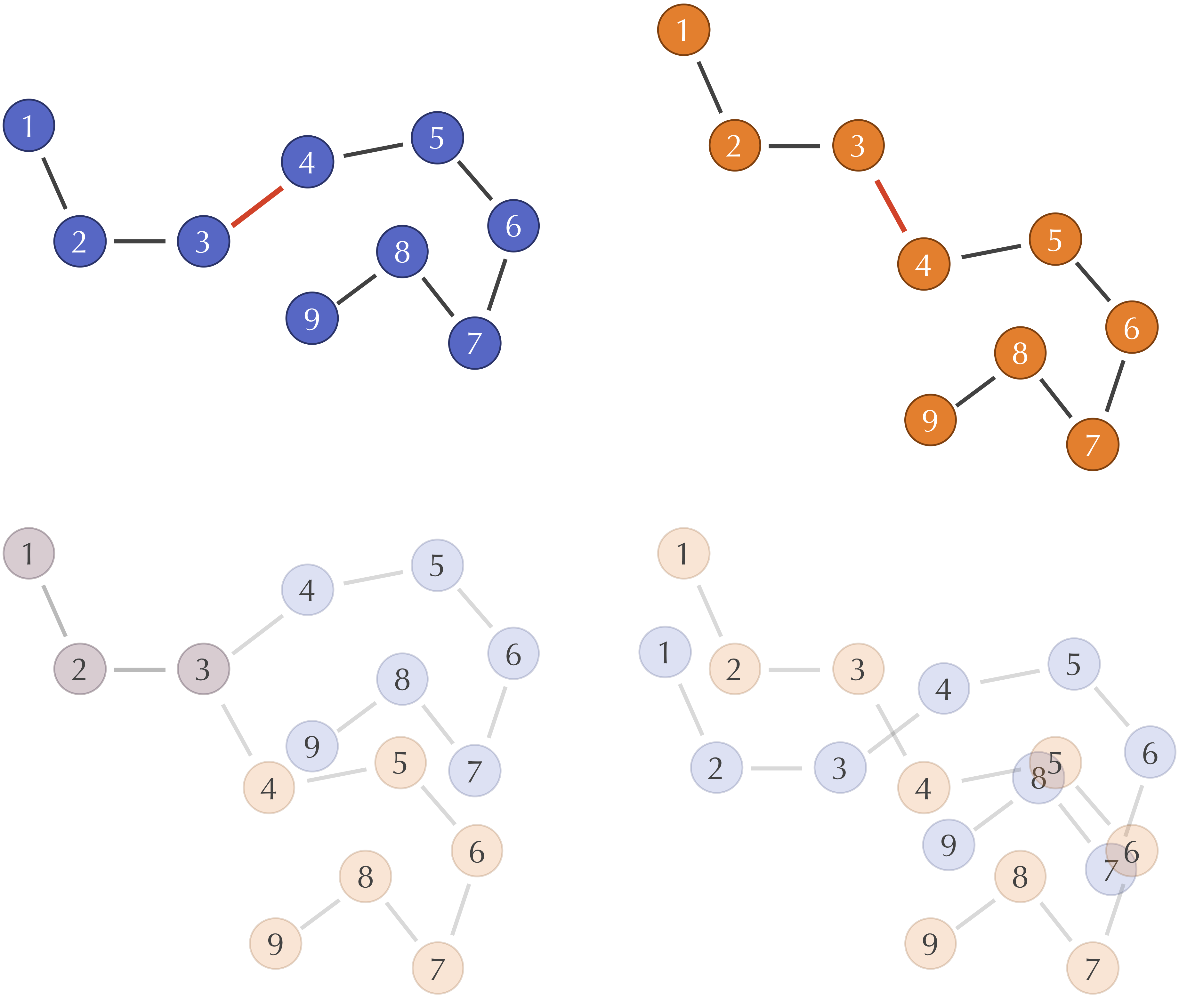
Learn to align molecular structures and point clouds using the Kabsch algorithm, with differentiable implementations for modern ML frameworks.