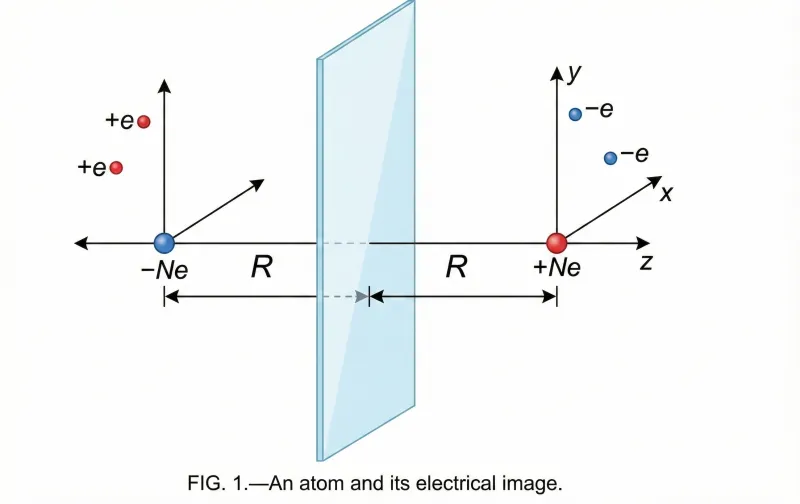
NInChI: Toward a Chemical Identifier for Nanomaterials
Can we create a SMILES-like notation for nanomaterials? A collaborative workshop tackles the challenge of representing complex, multi-component nanomaterials with a proposed extension to the established InChI system.