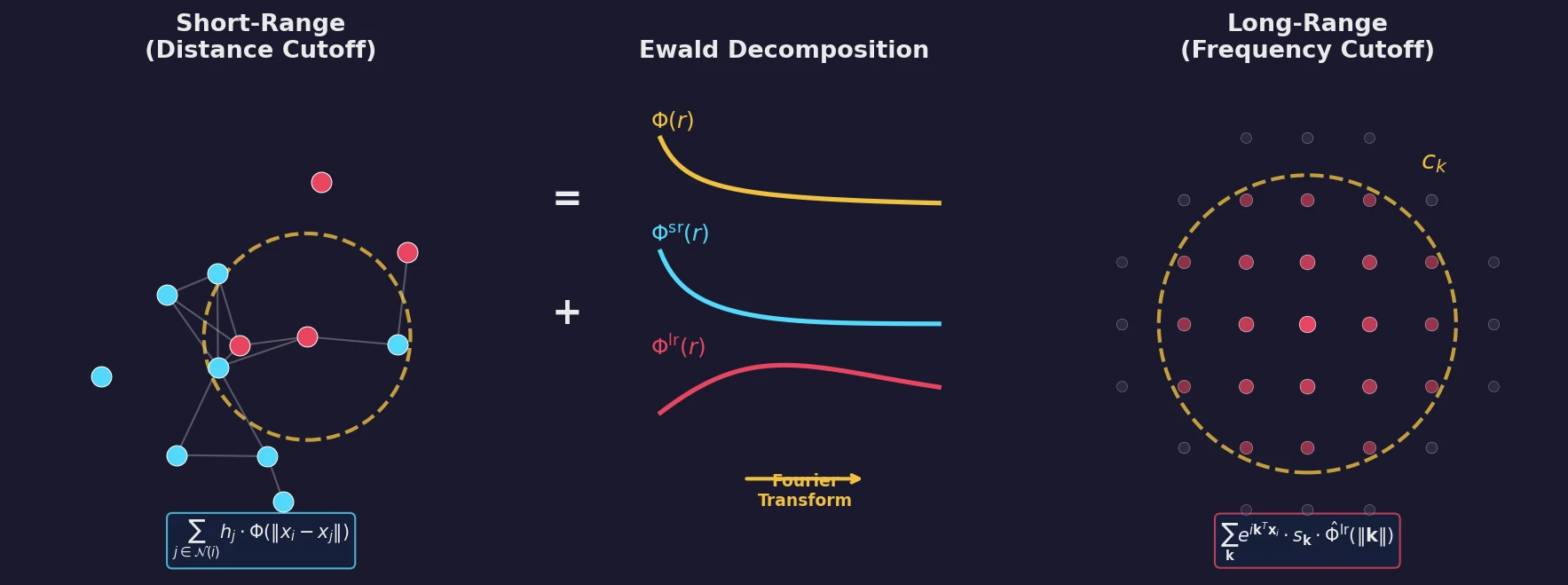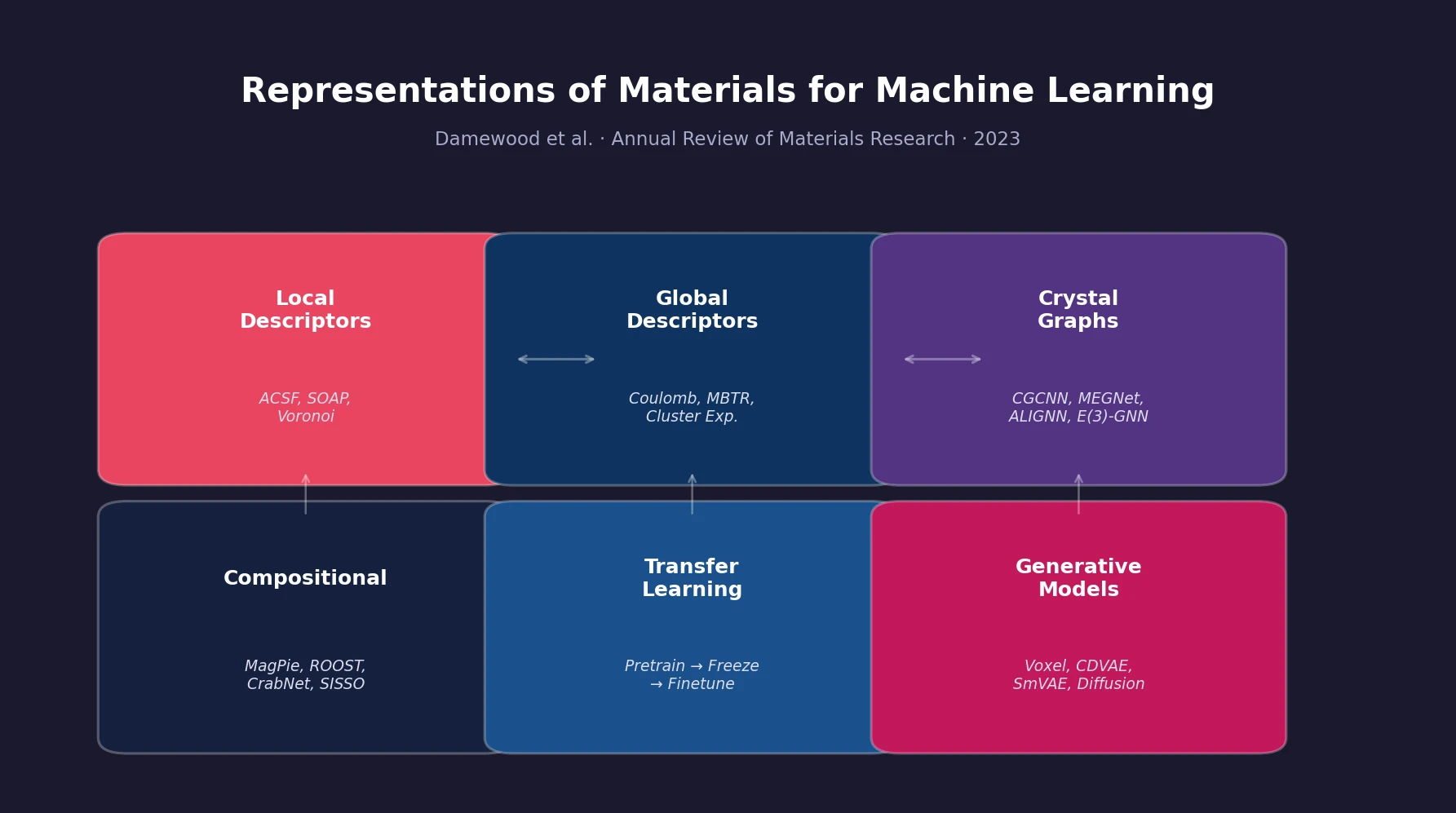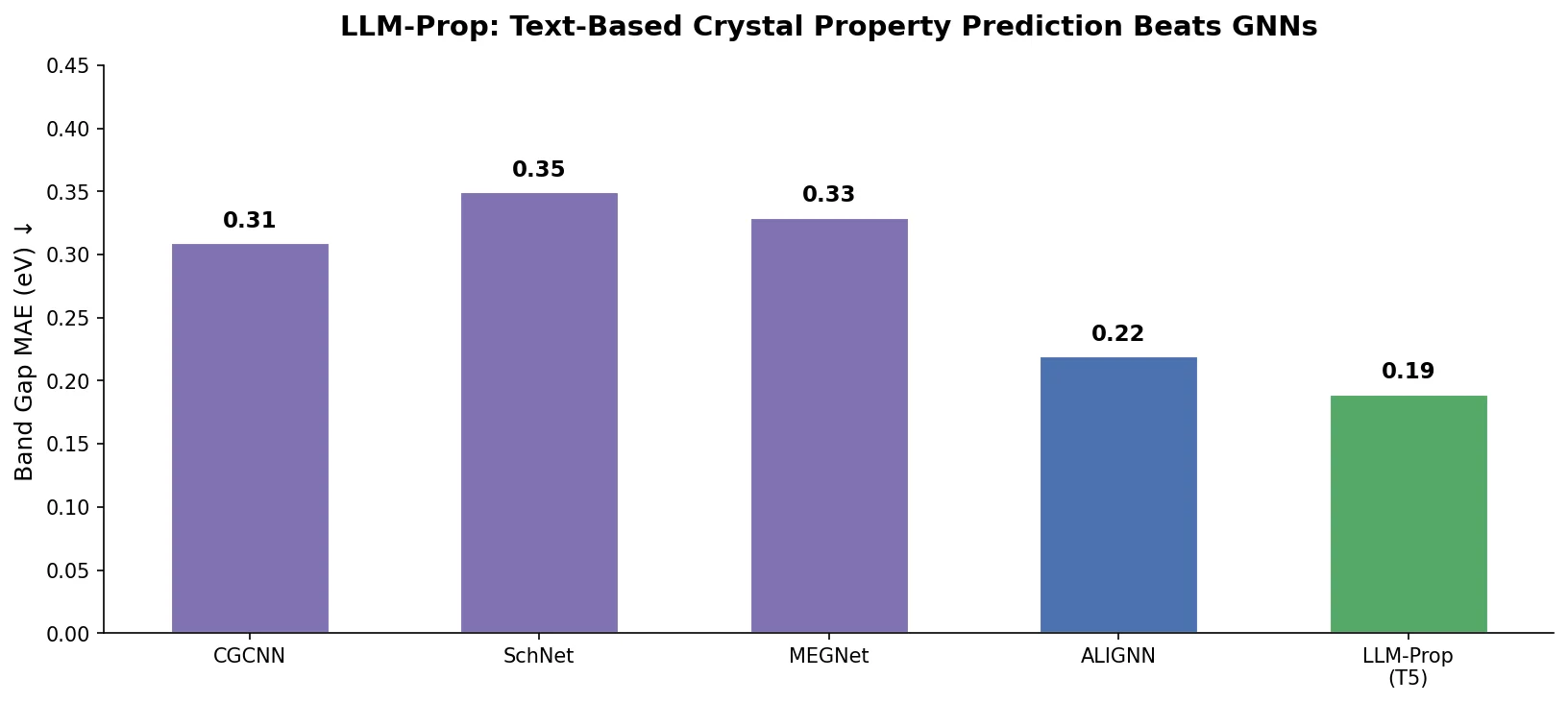
Atom-Density Representations for Machine Learning
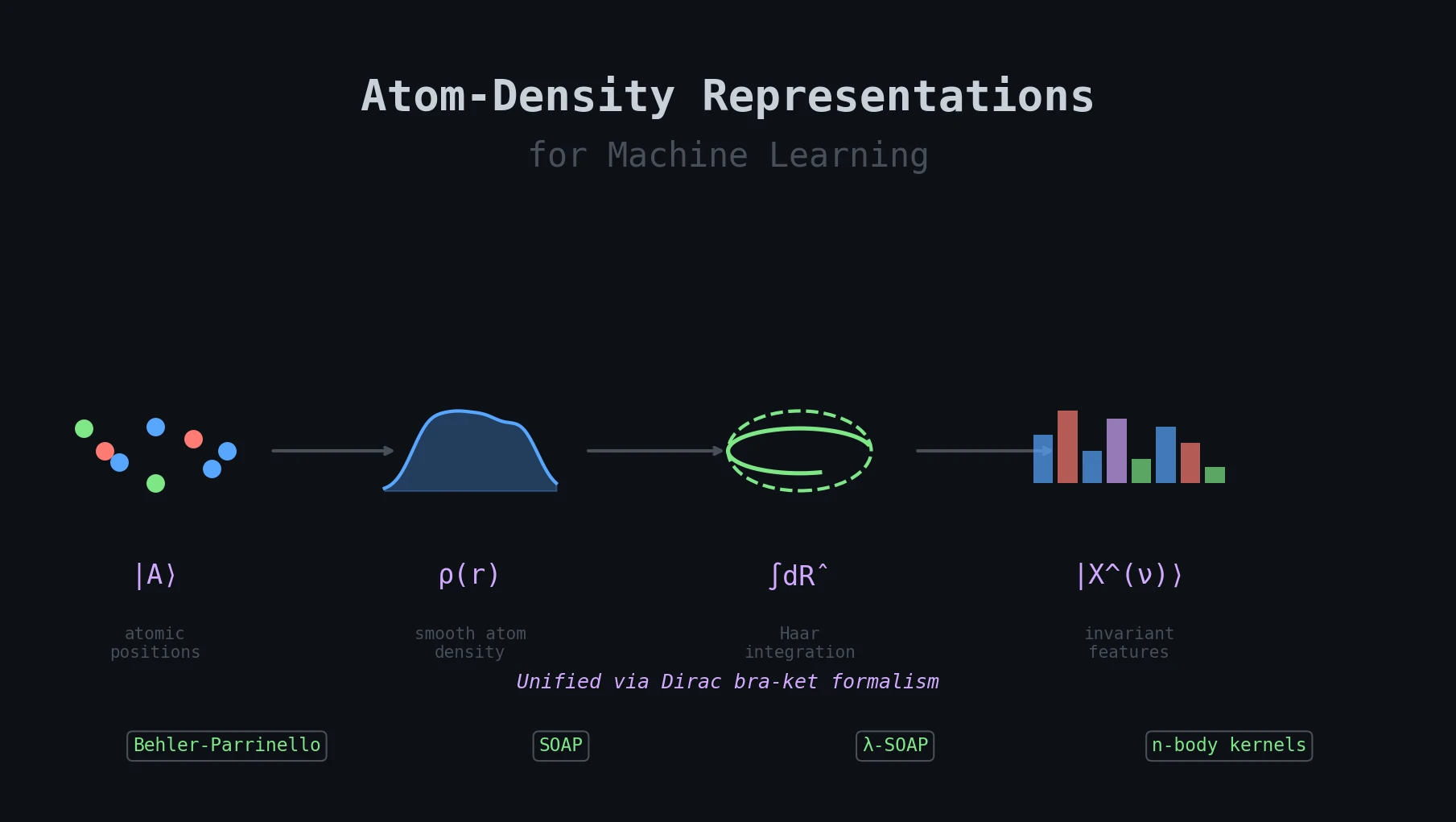
Introduces a Dirac notation formalism for atomic environments that unifies SOAP power spectra, Behler-Parrinello symmetry functions, and other density-based structural representations under a single theoretical framework.