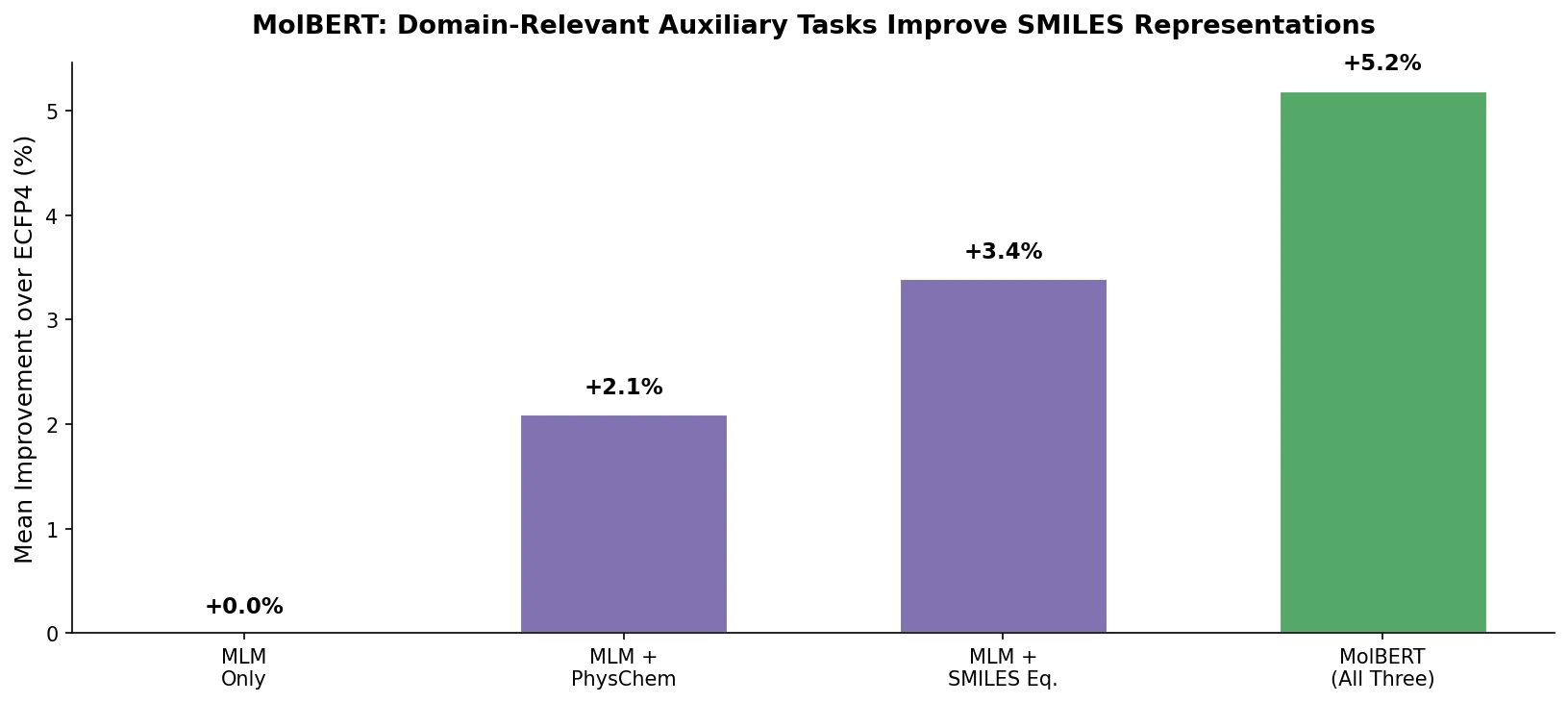
MolBERT: Auxiliary Tasks for Molecular BERT Models
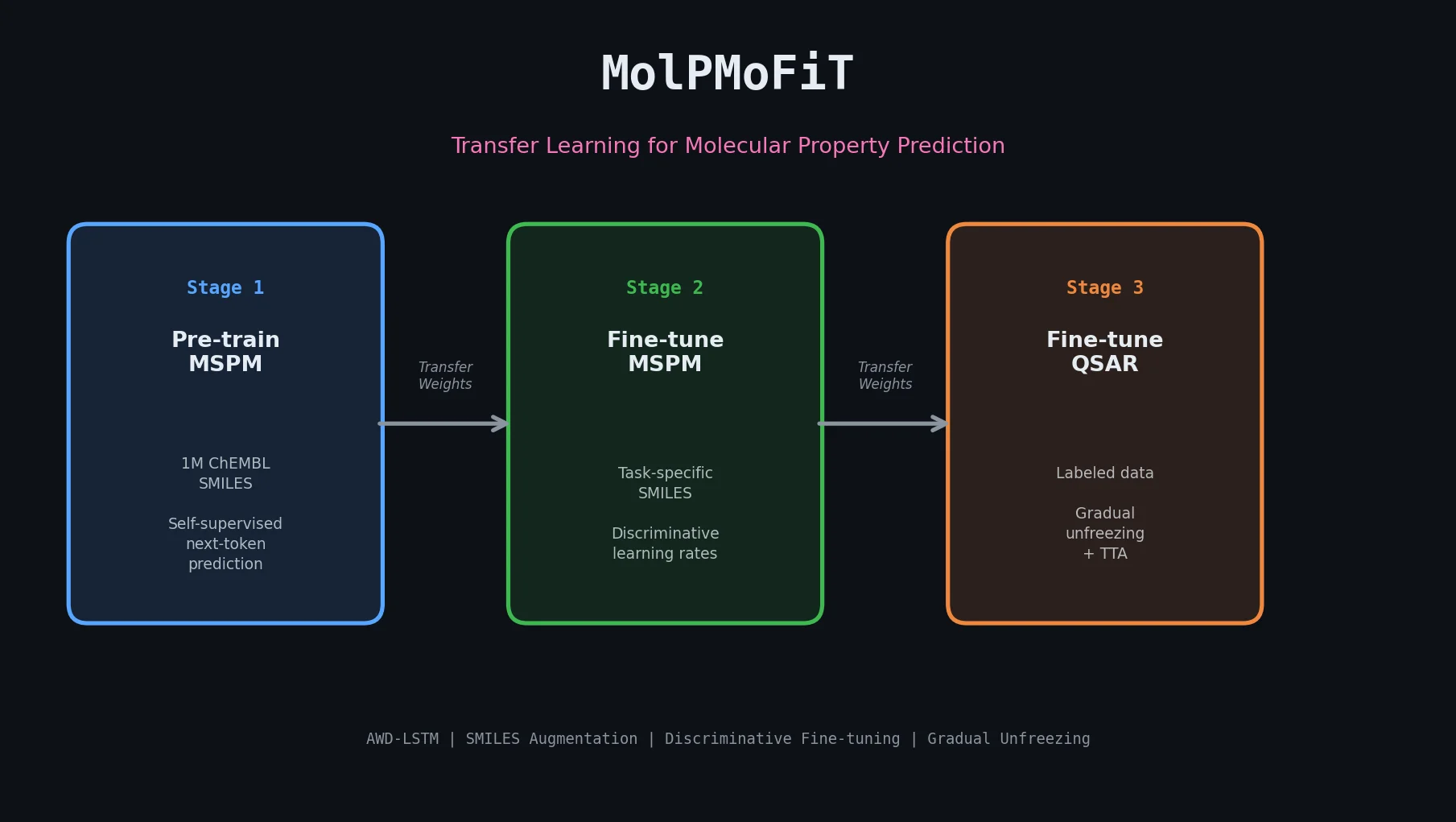
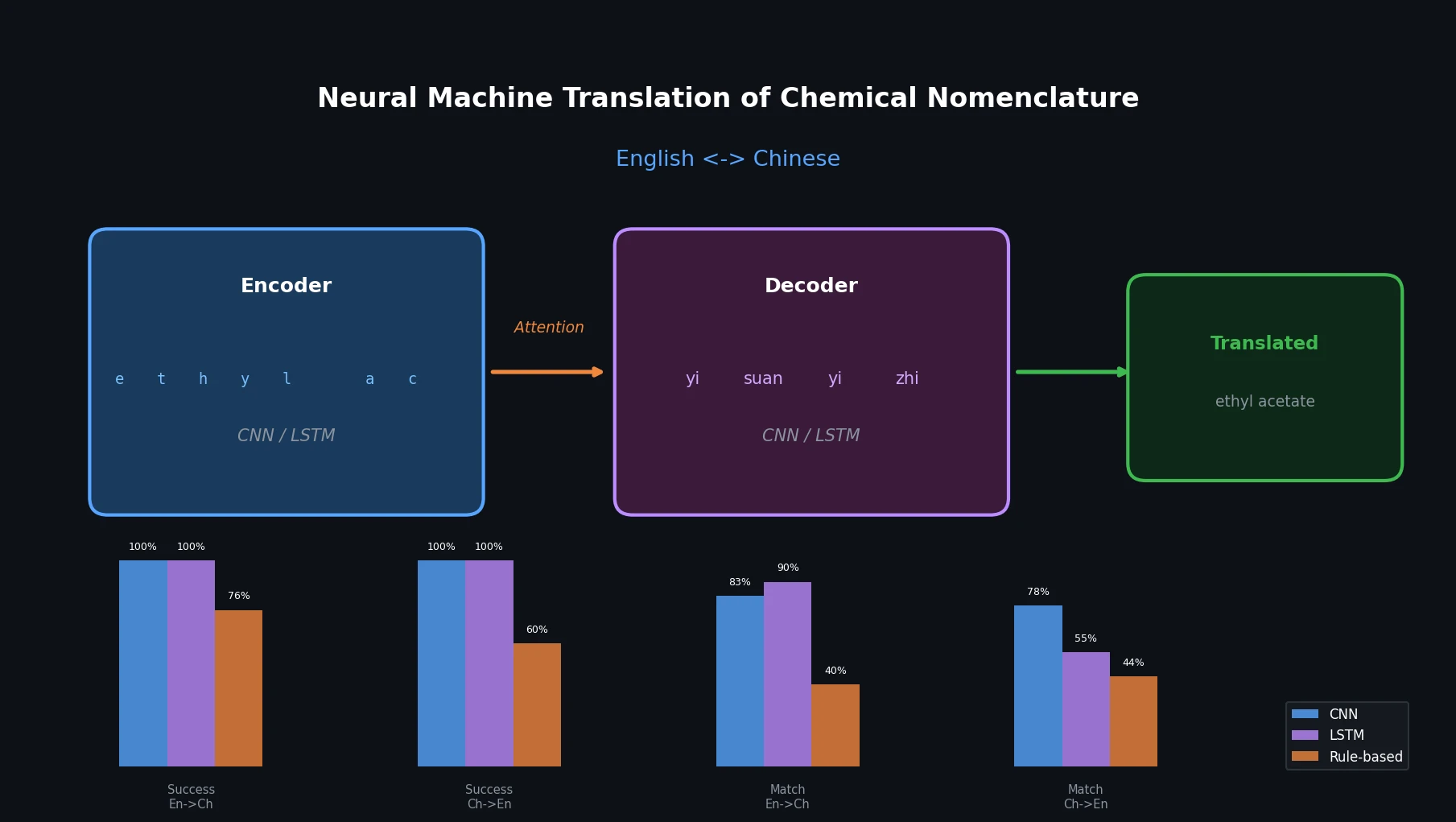
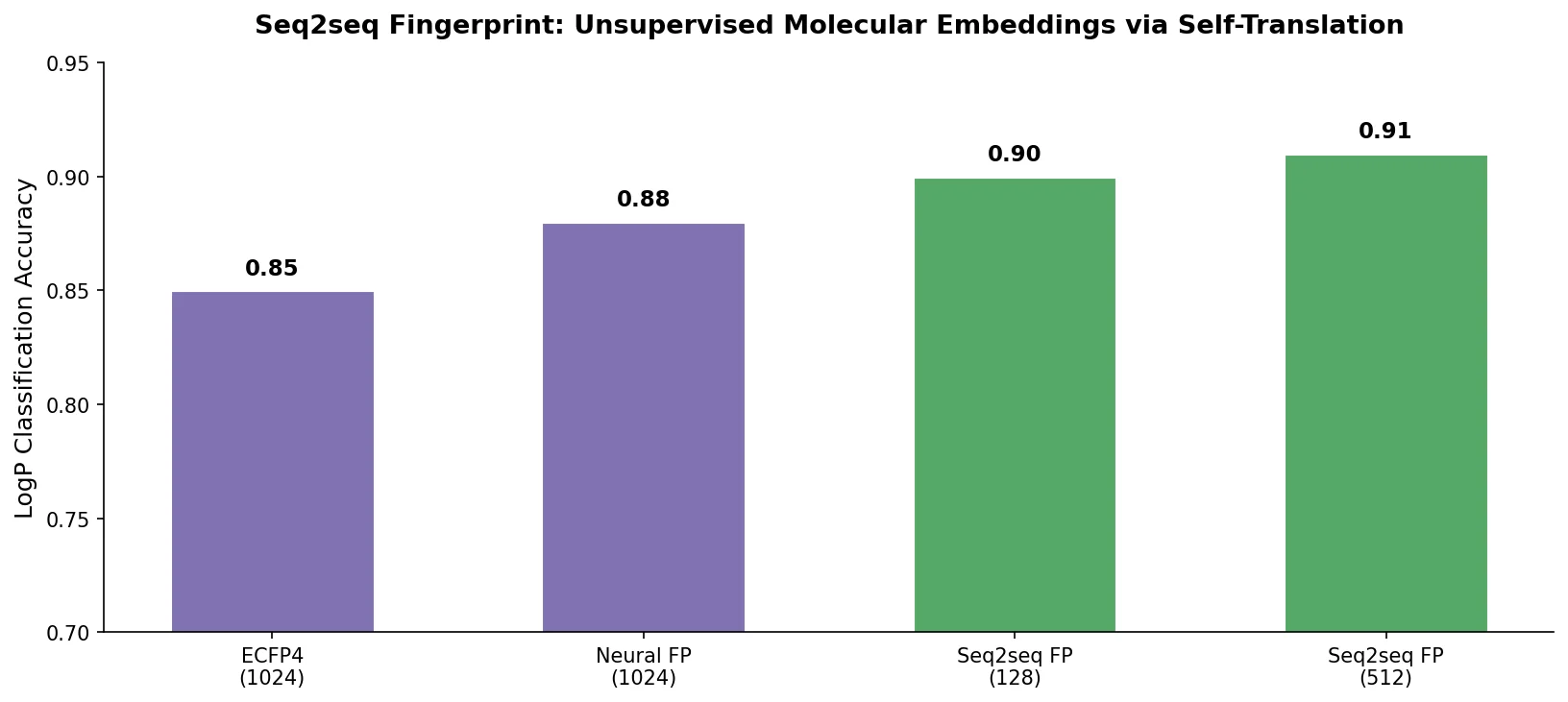
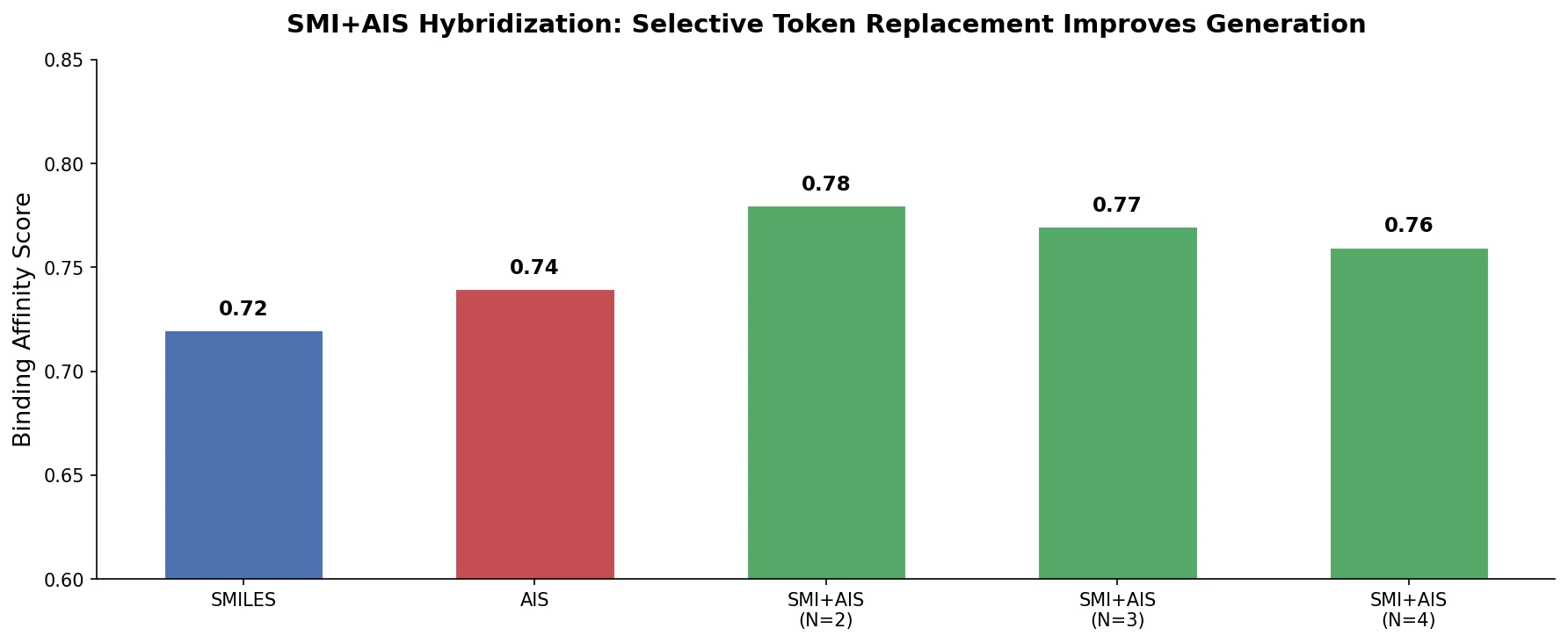
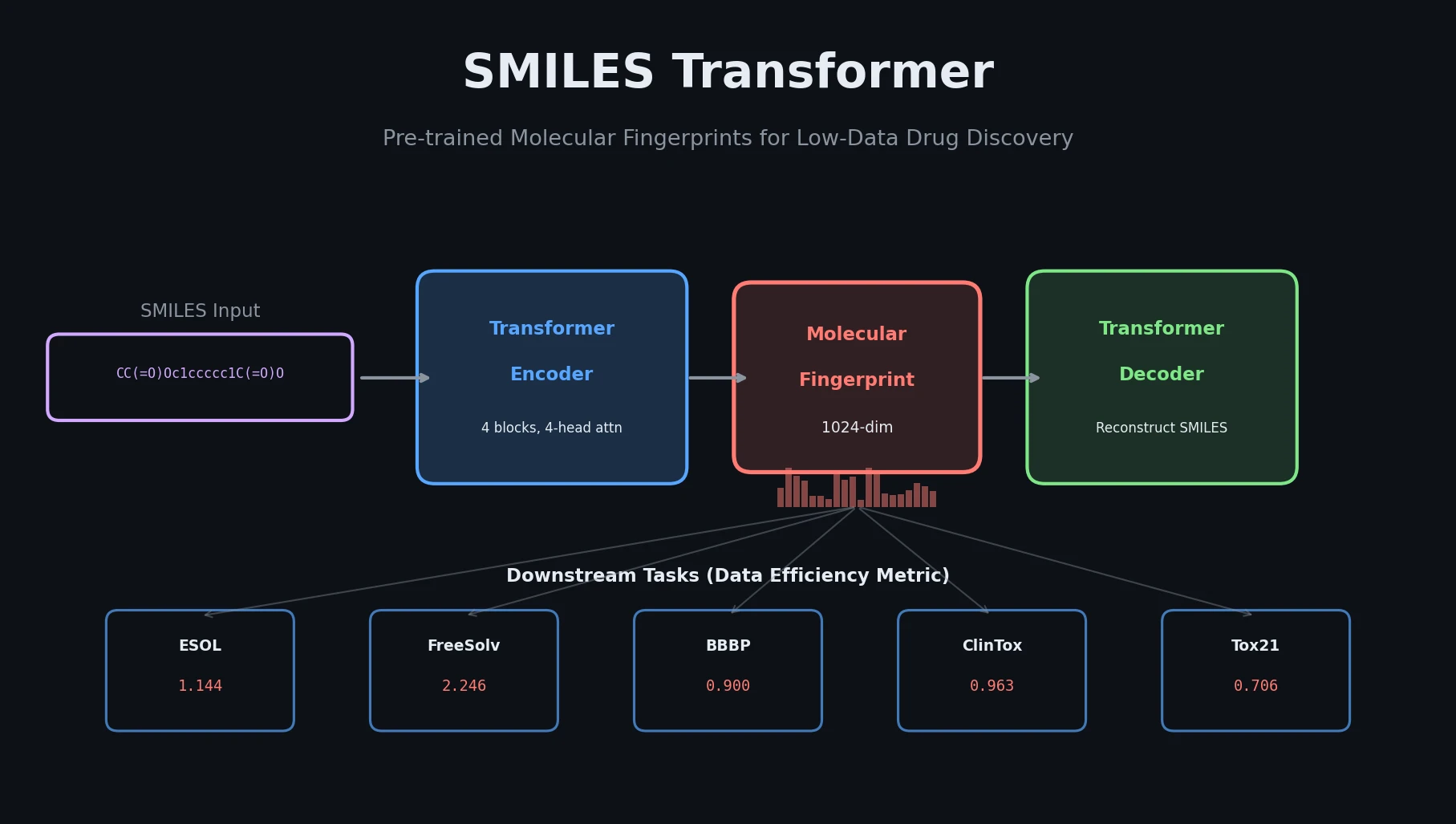
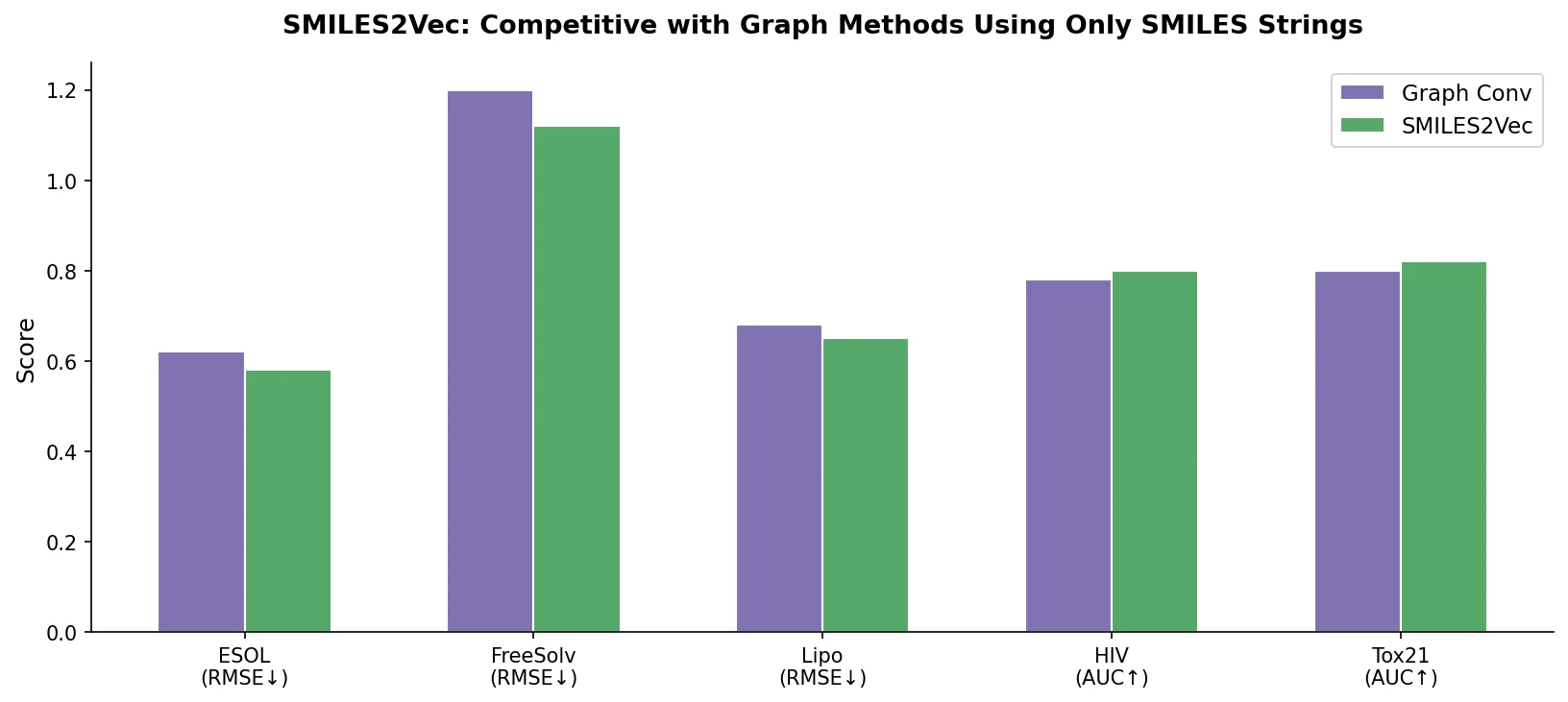
MolBERT pre-trains a BERT model on SMILES strings using masked language modeling, SMILES equivalence, and physicochemical property prediction as auxiliary tasks, achieving state-of-the-art results on virtual screening and QSAR benchmarks.