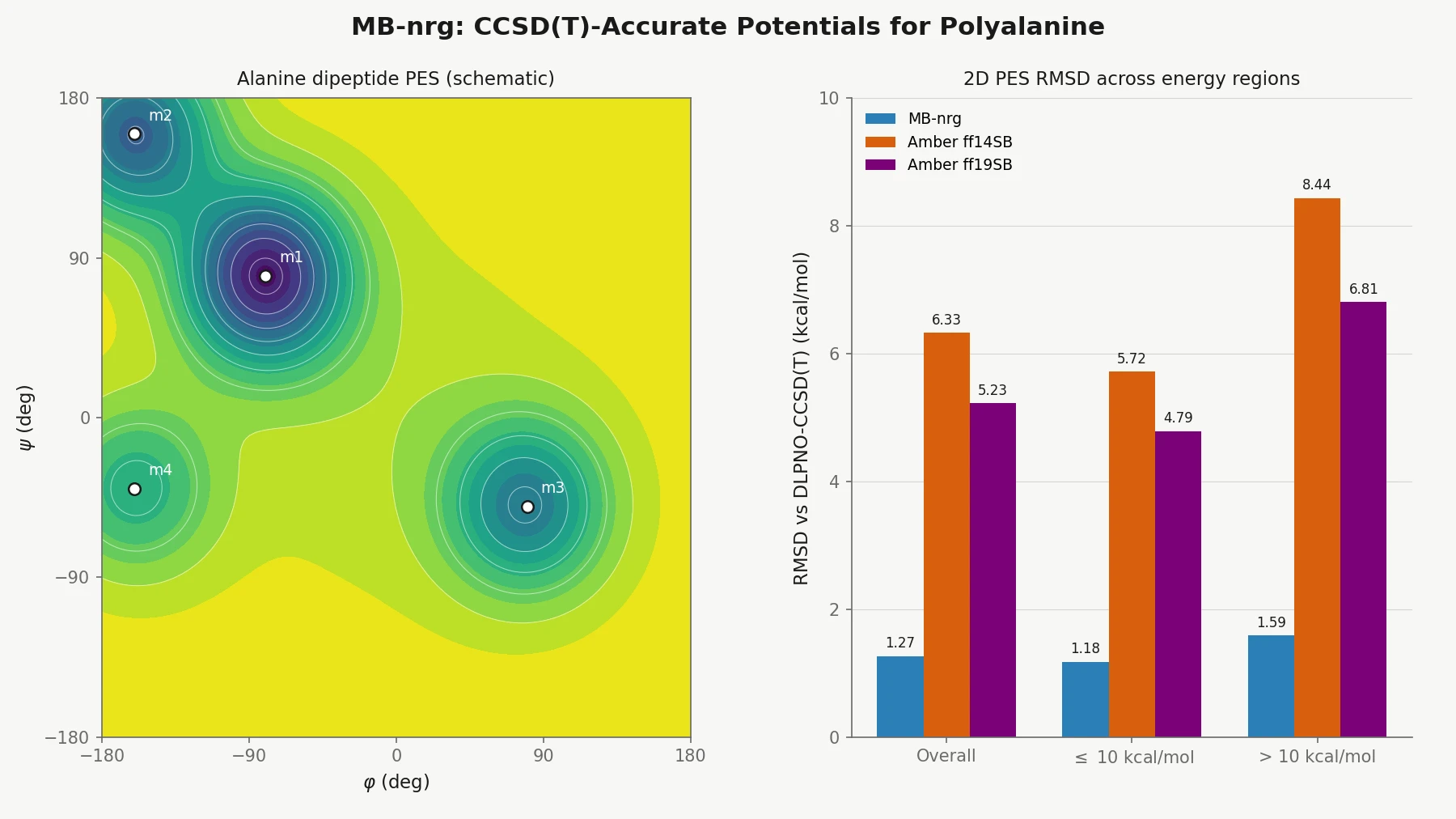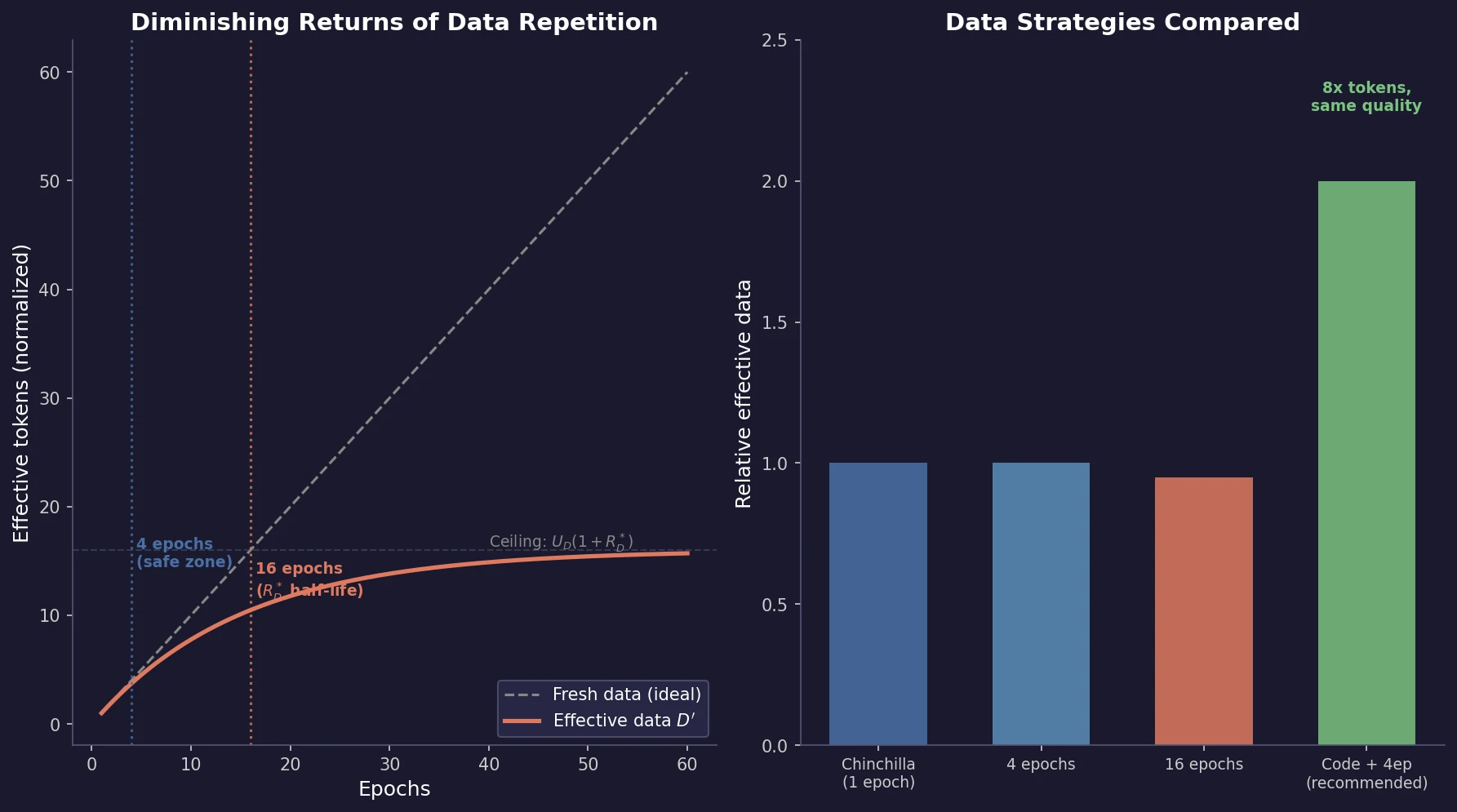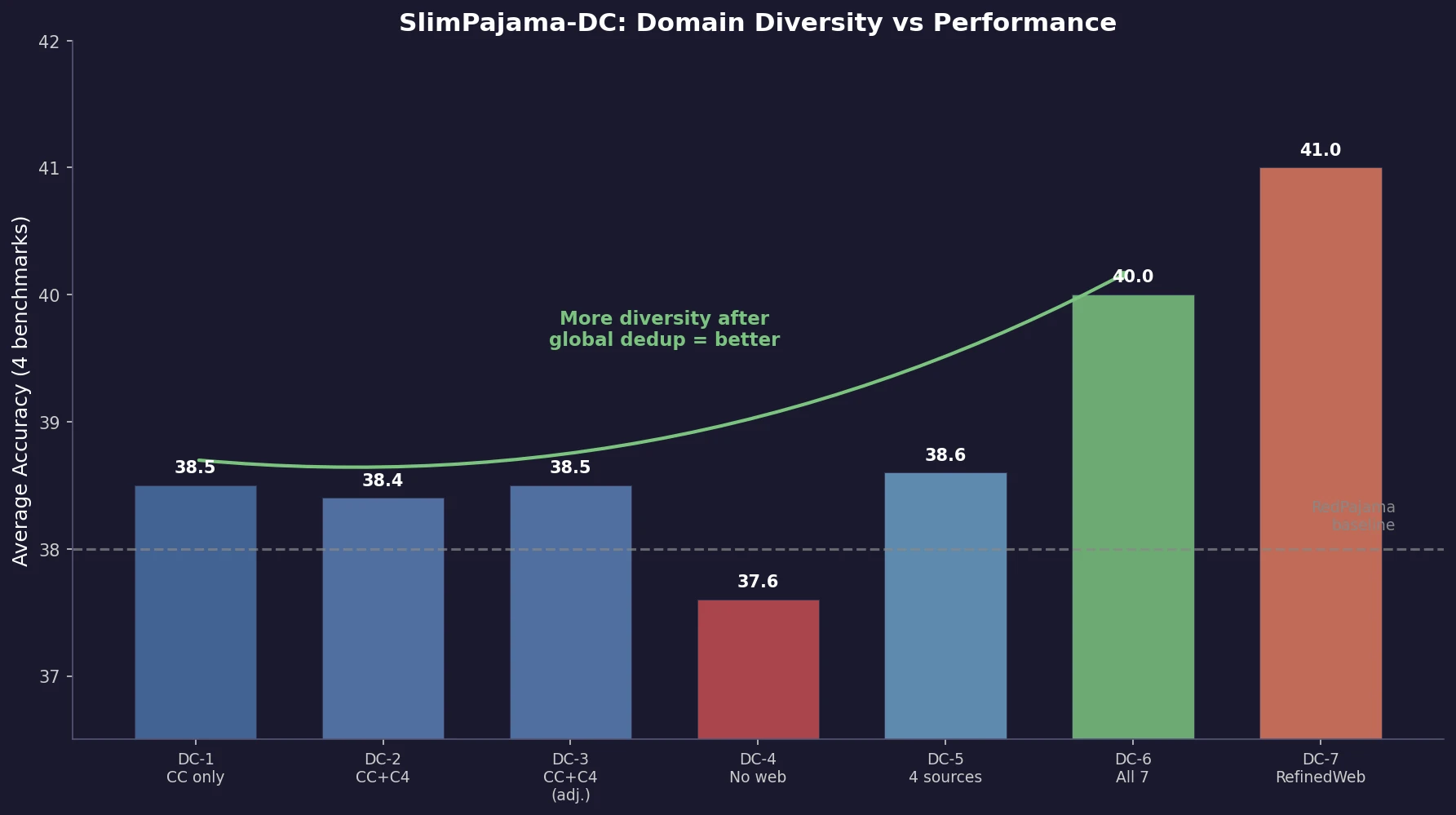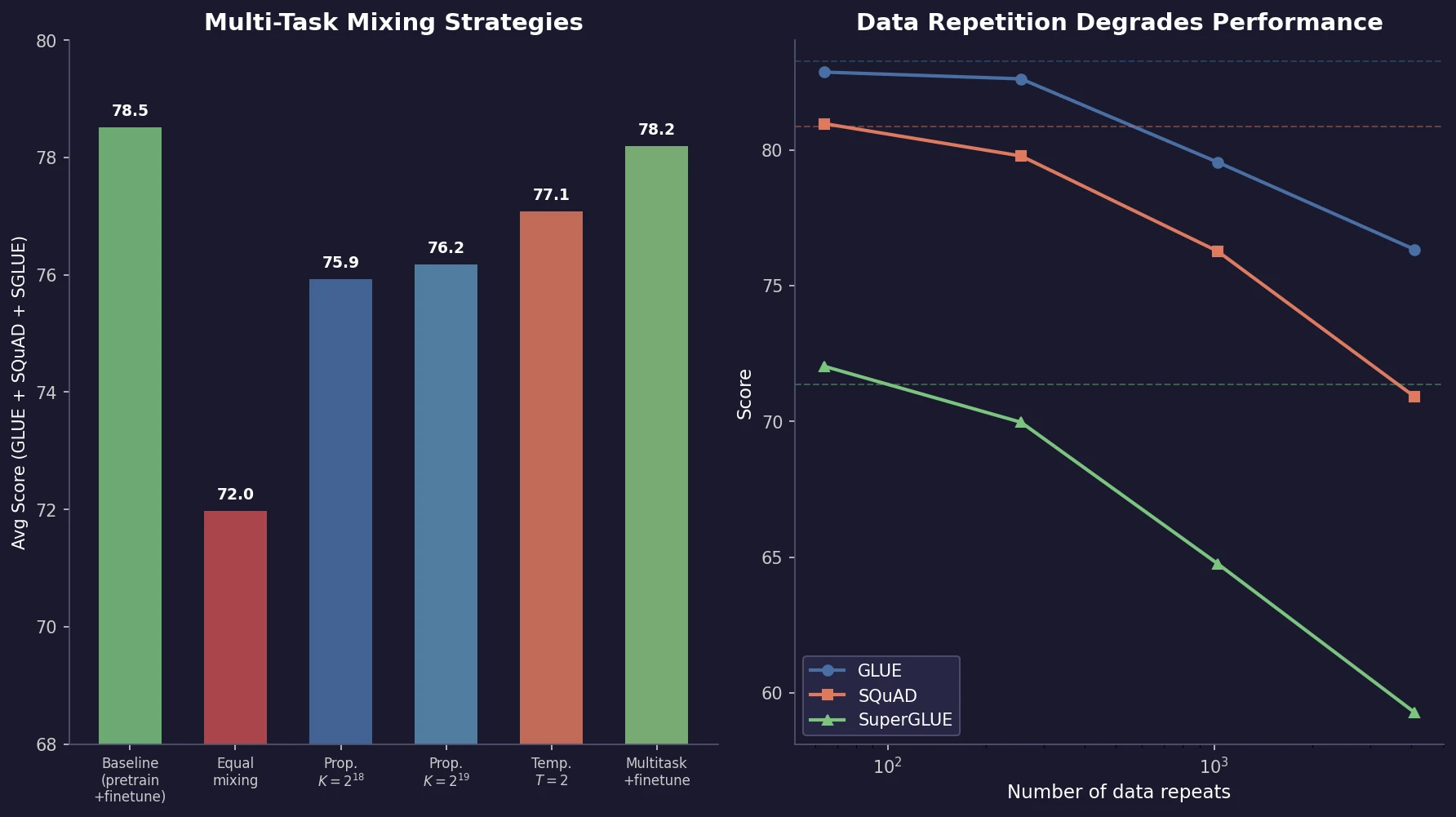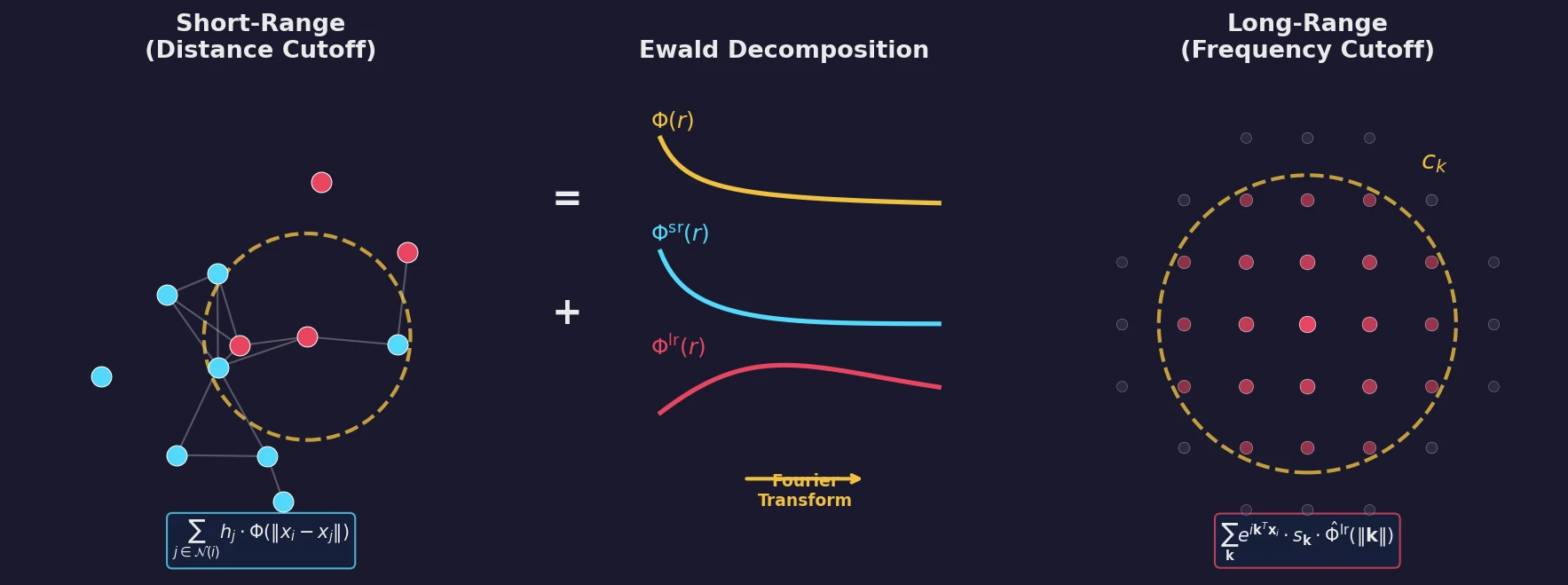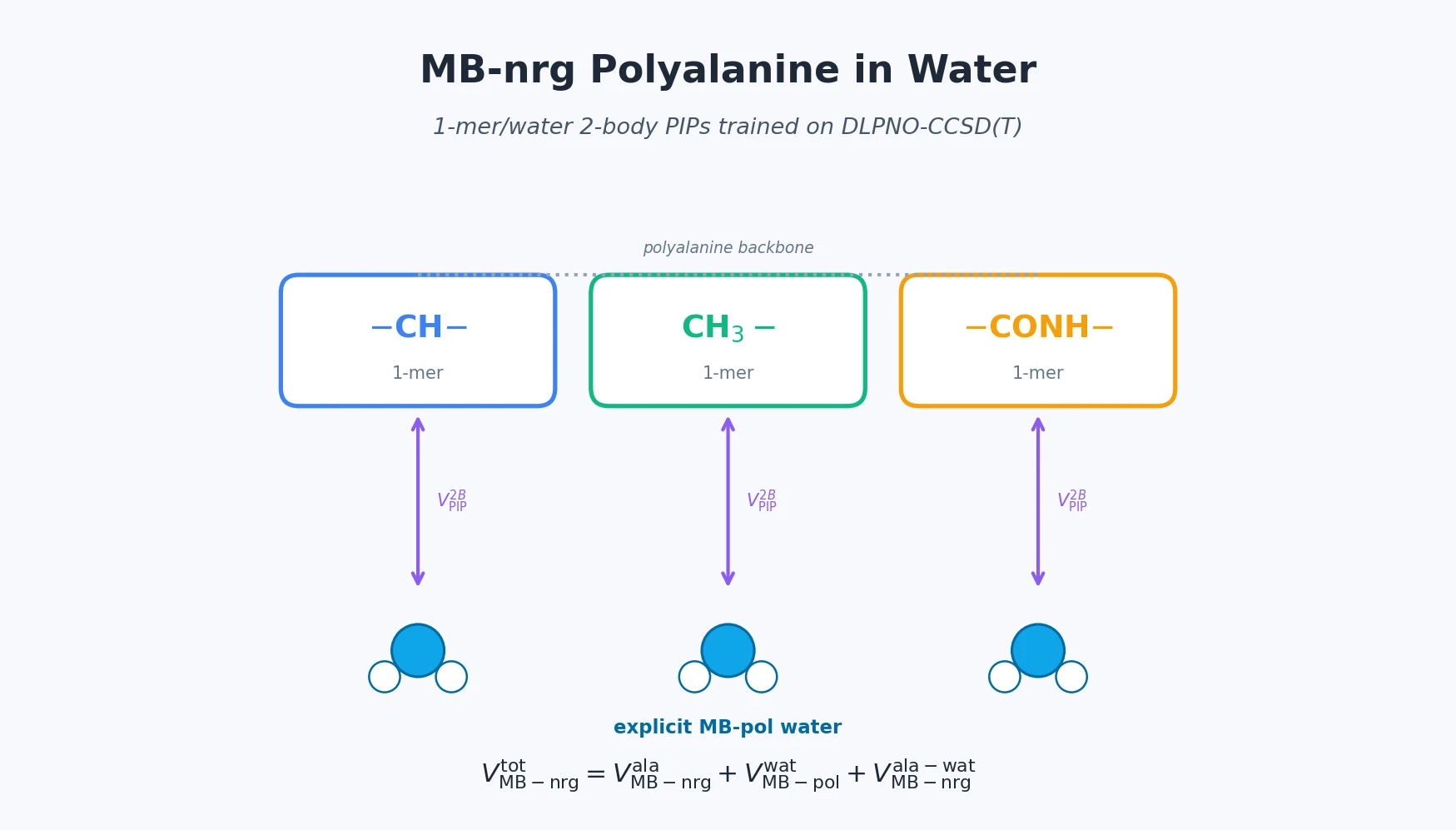
MB-nrg in Solution: Polyalanine in Water with CCSD(T) PEFs
Building on the gas-phase MB-nrg PEF for polyalanine, Ruihan Zhou and Francesco Paesani add machine-learned 2-body terms for each backbone functional group interacting with water, fit to BSSE-corrected DLPNO-CCSD(T)/aug-cc-pVTZ data, then validate the resulting potential against alanine dipeptide-water dimer scans, free-energy surfaces in explicit MB-pol water, and hydration radial distribution functions.