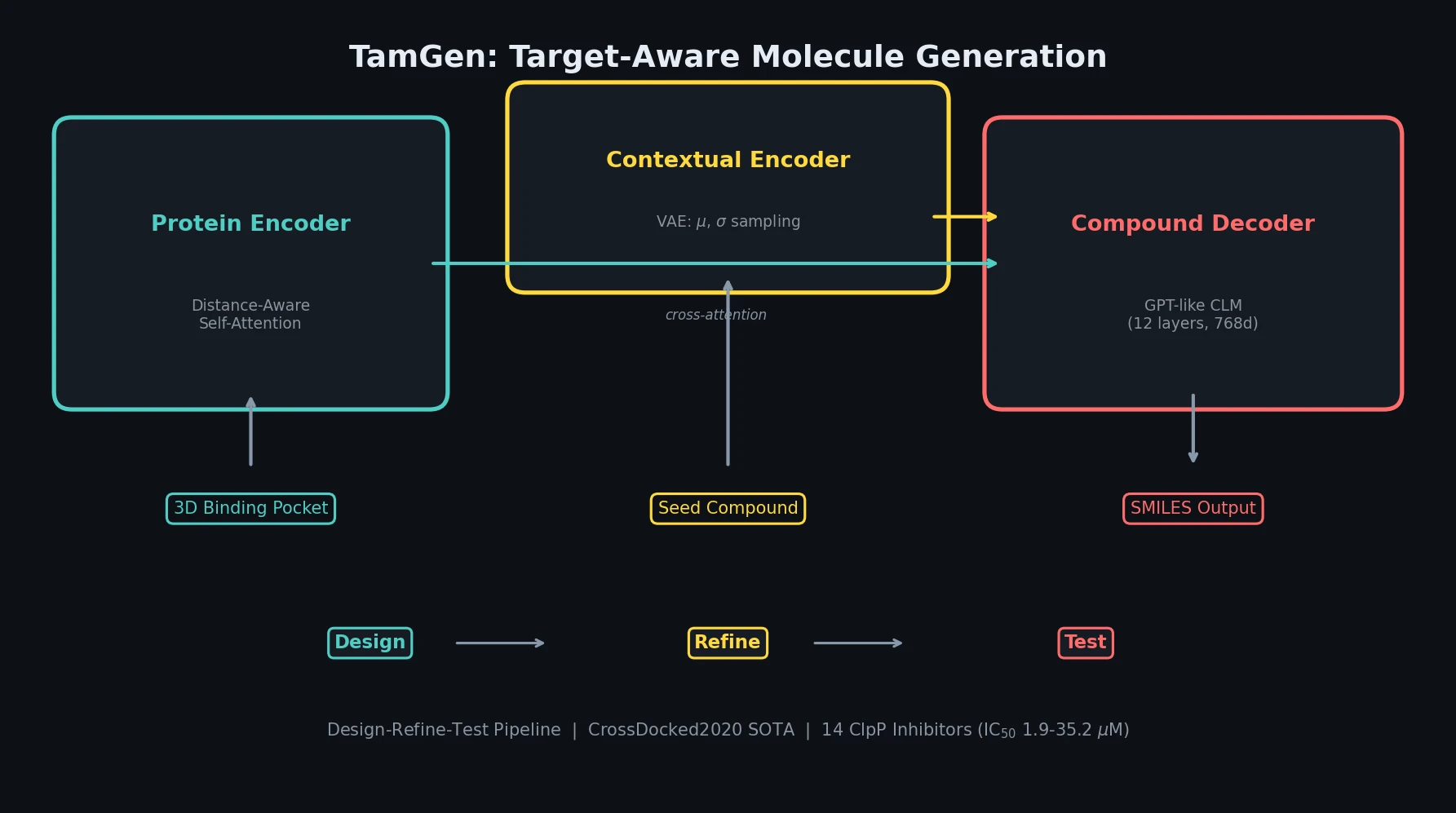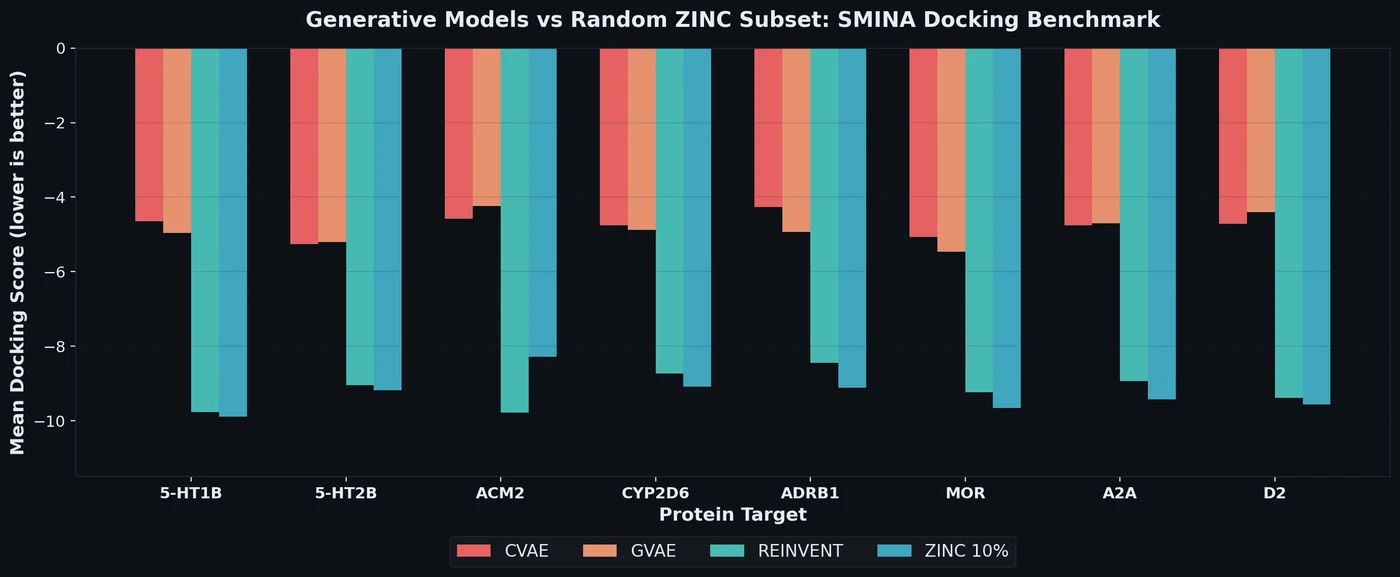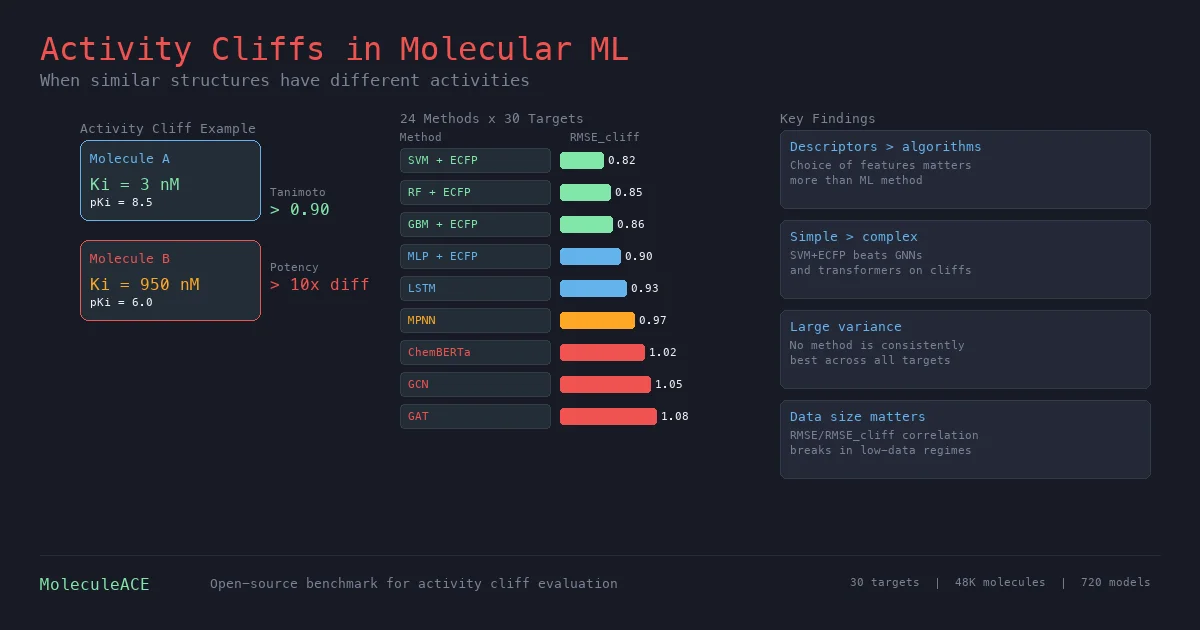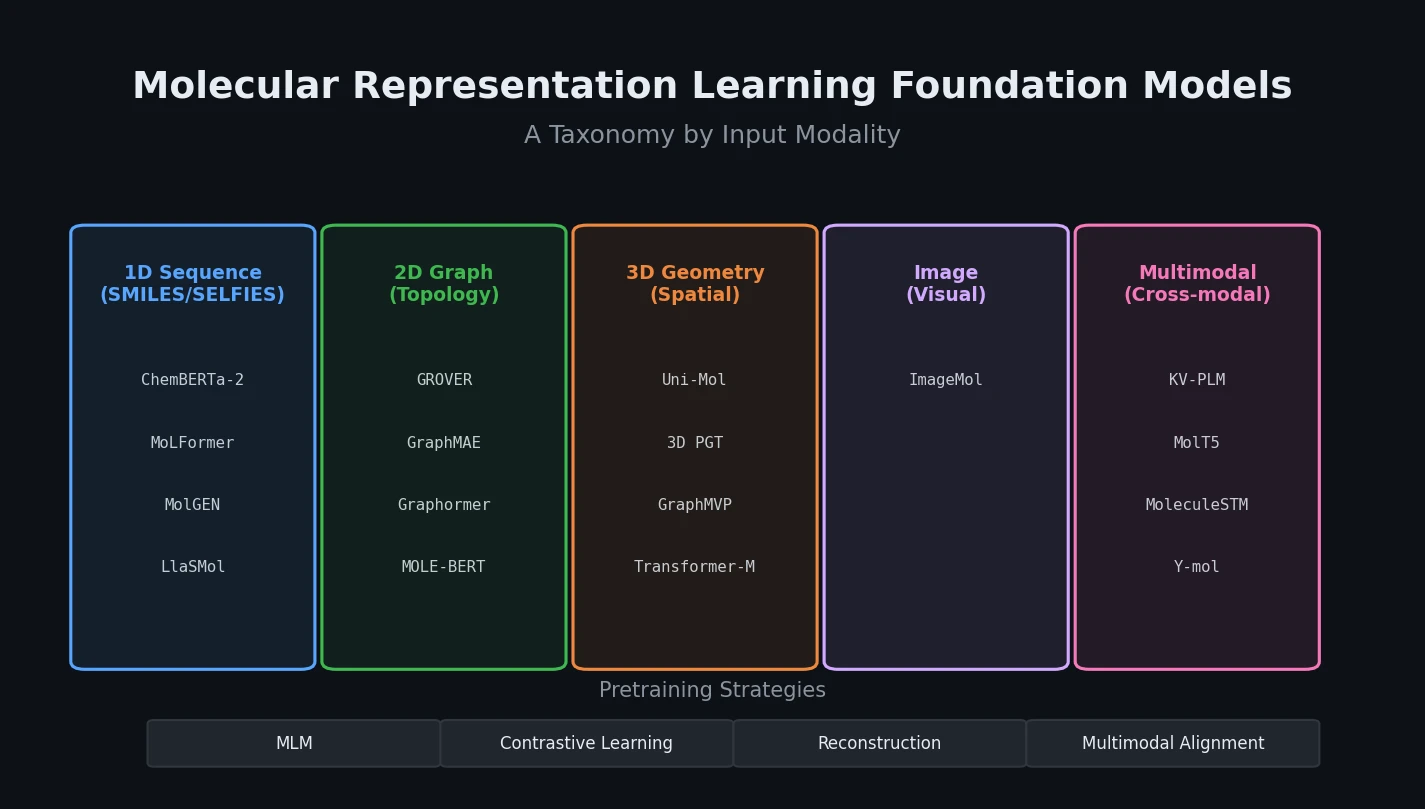
Review of Molecular Representation Learning Models
A comprehensive survey classifying molecular representation learning foundation models by input modality (sequence, graph, 3D, image, multimodal) and analyzing four pretraining paradigms for drug discovery tasks.