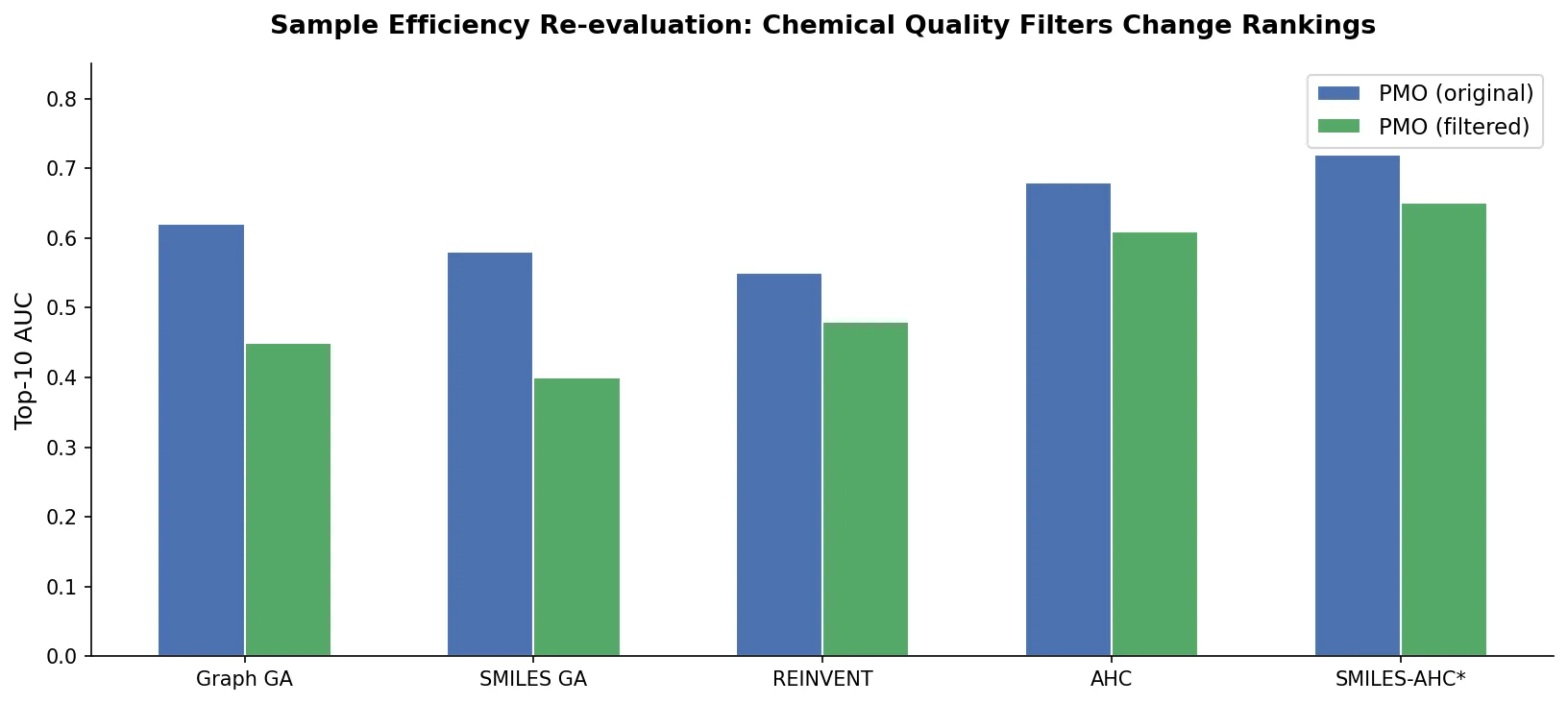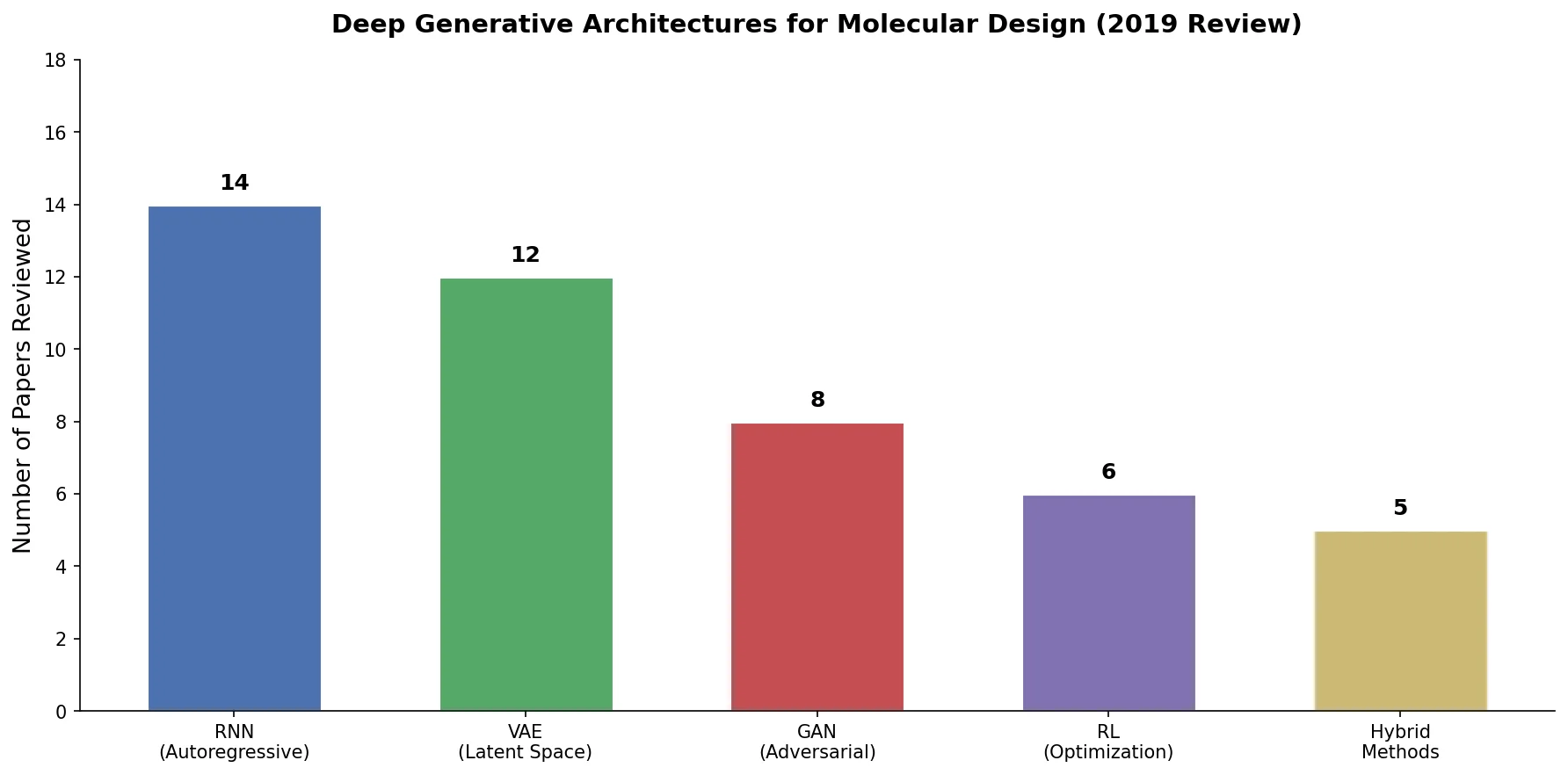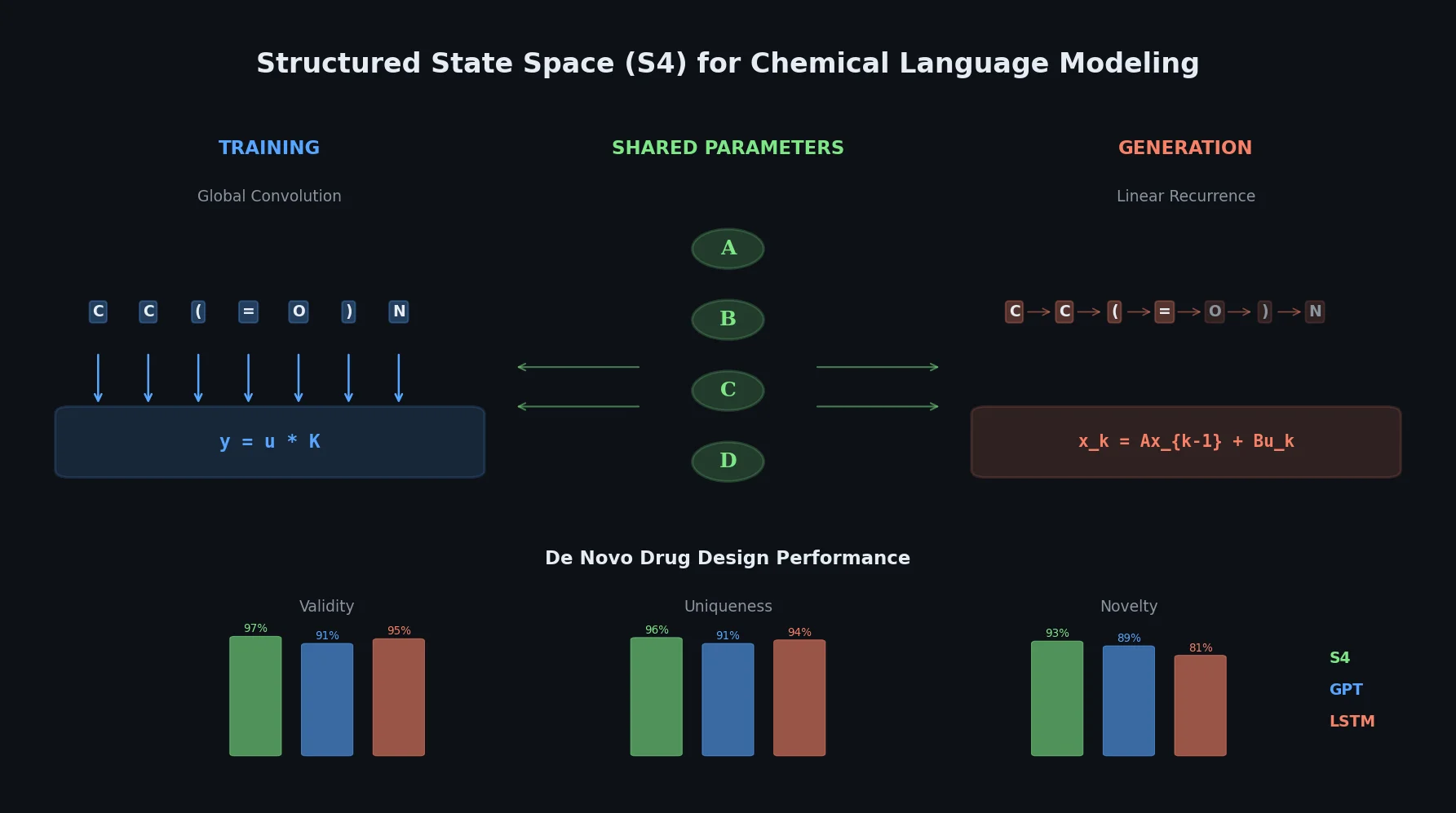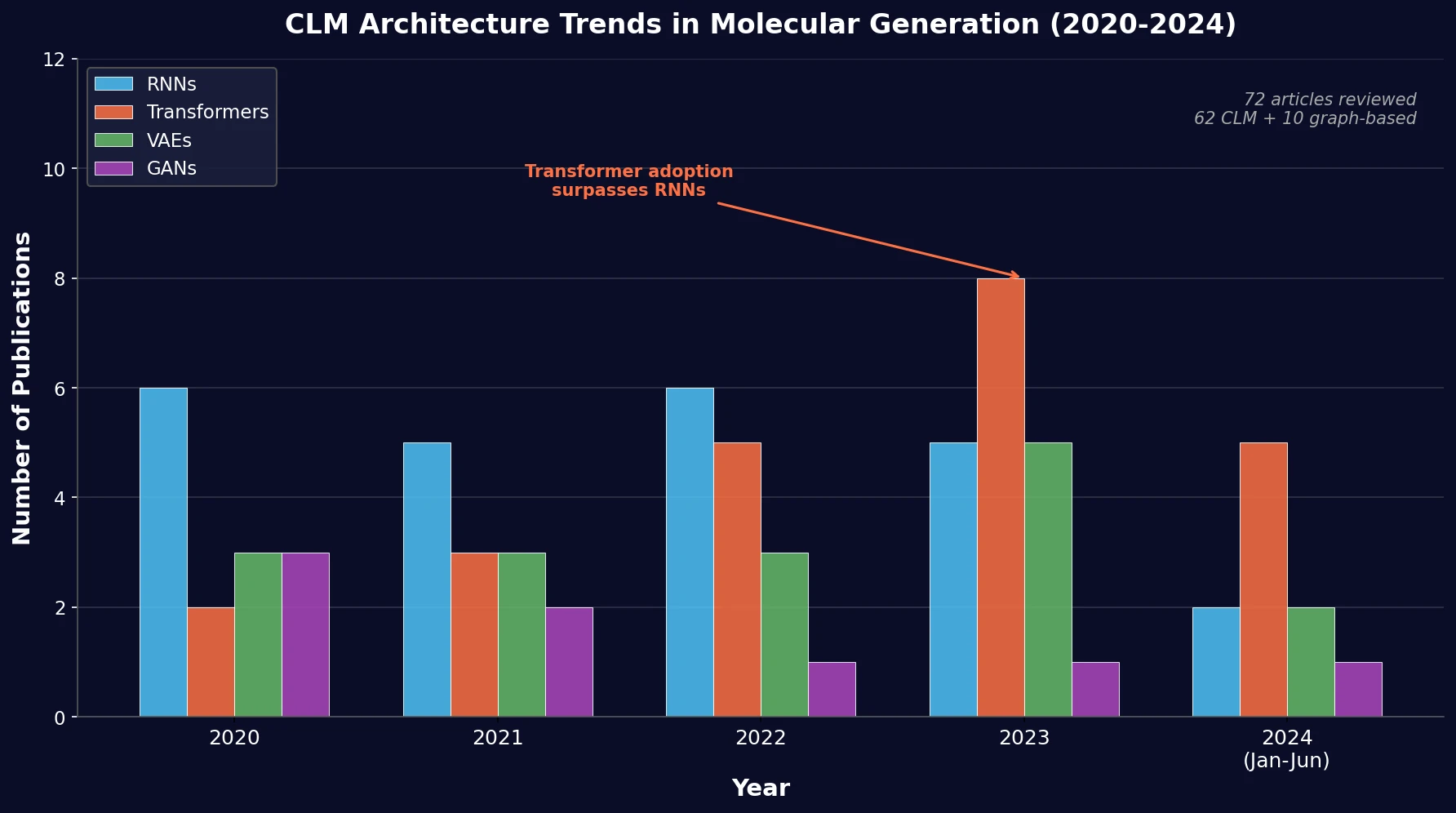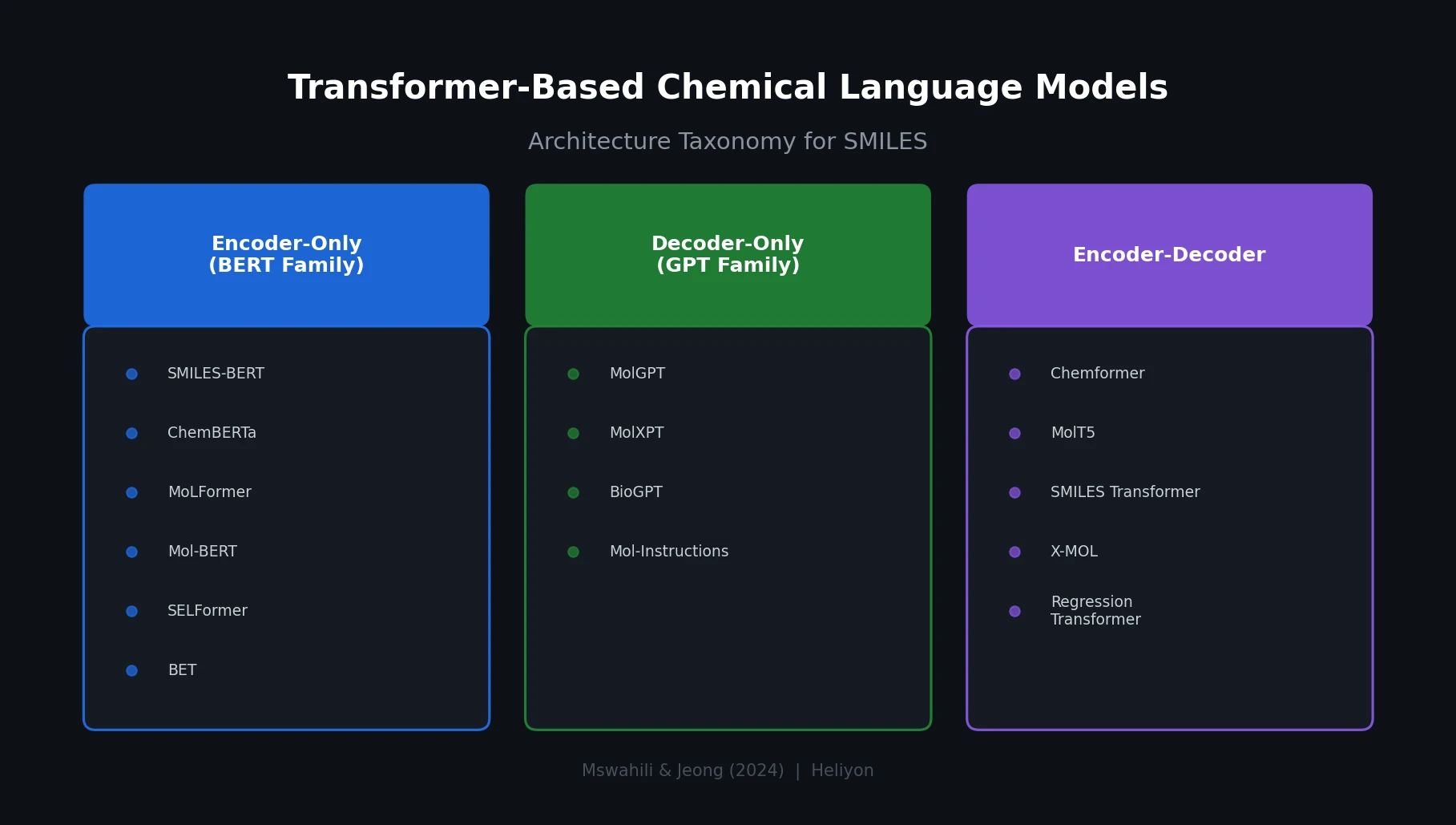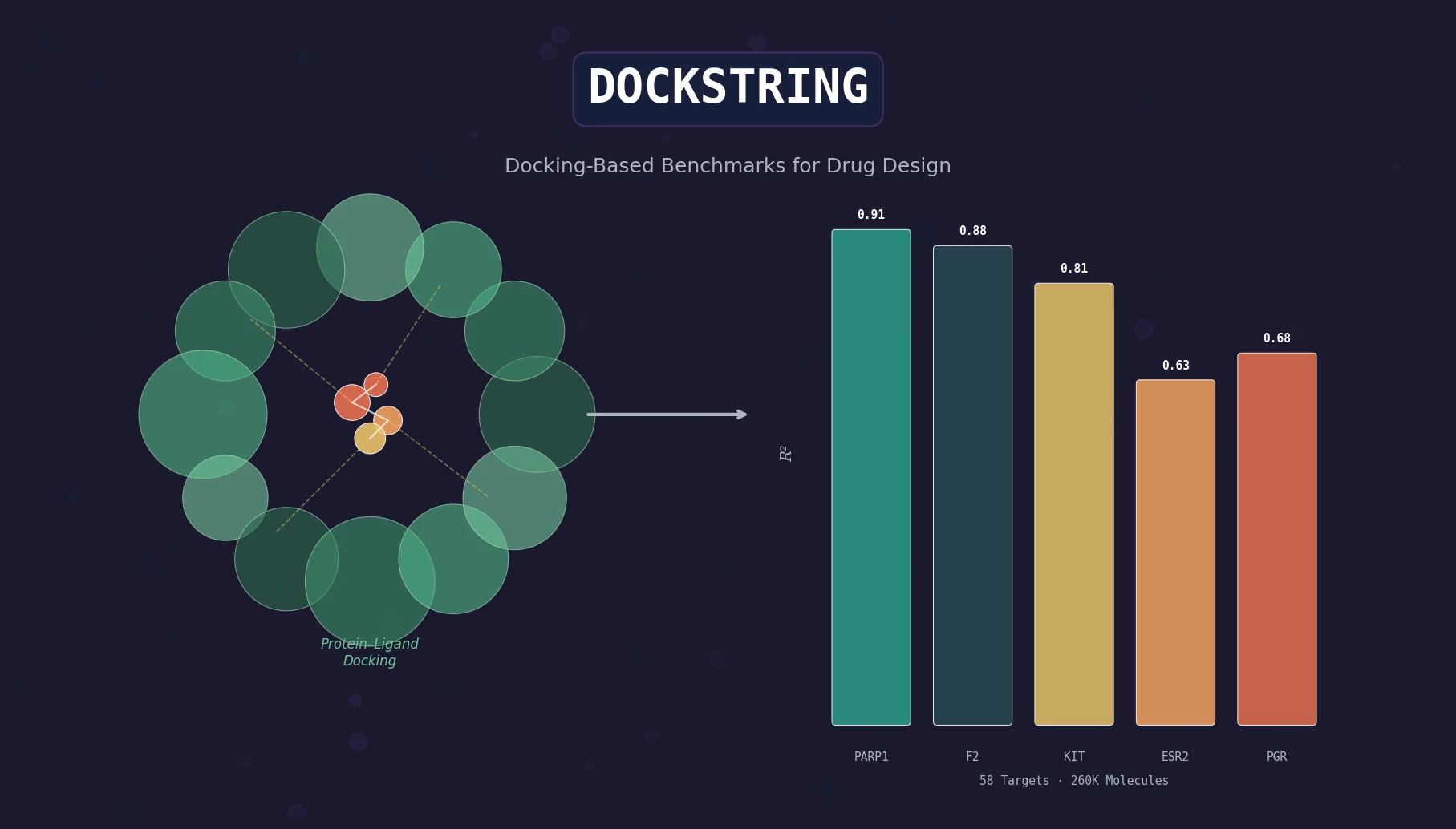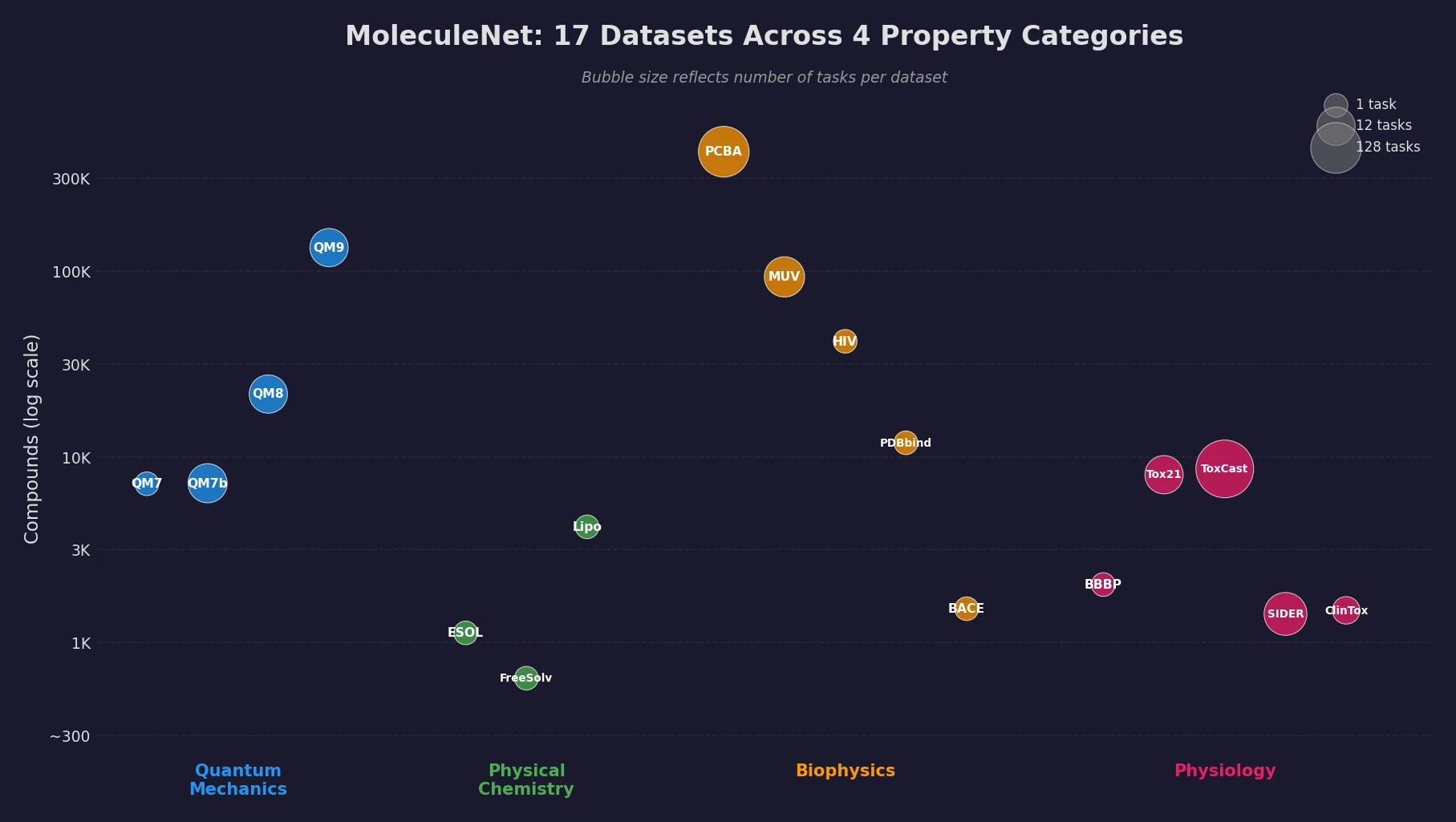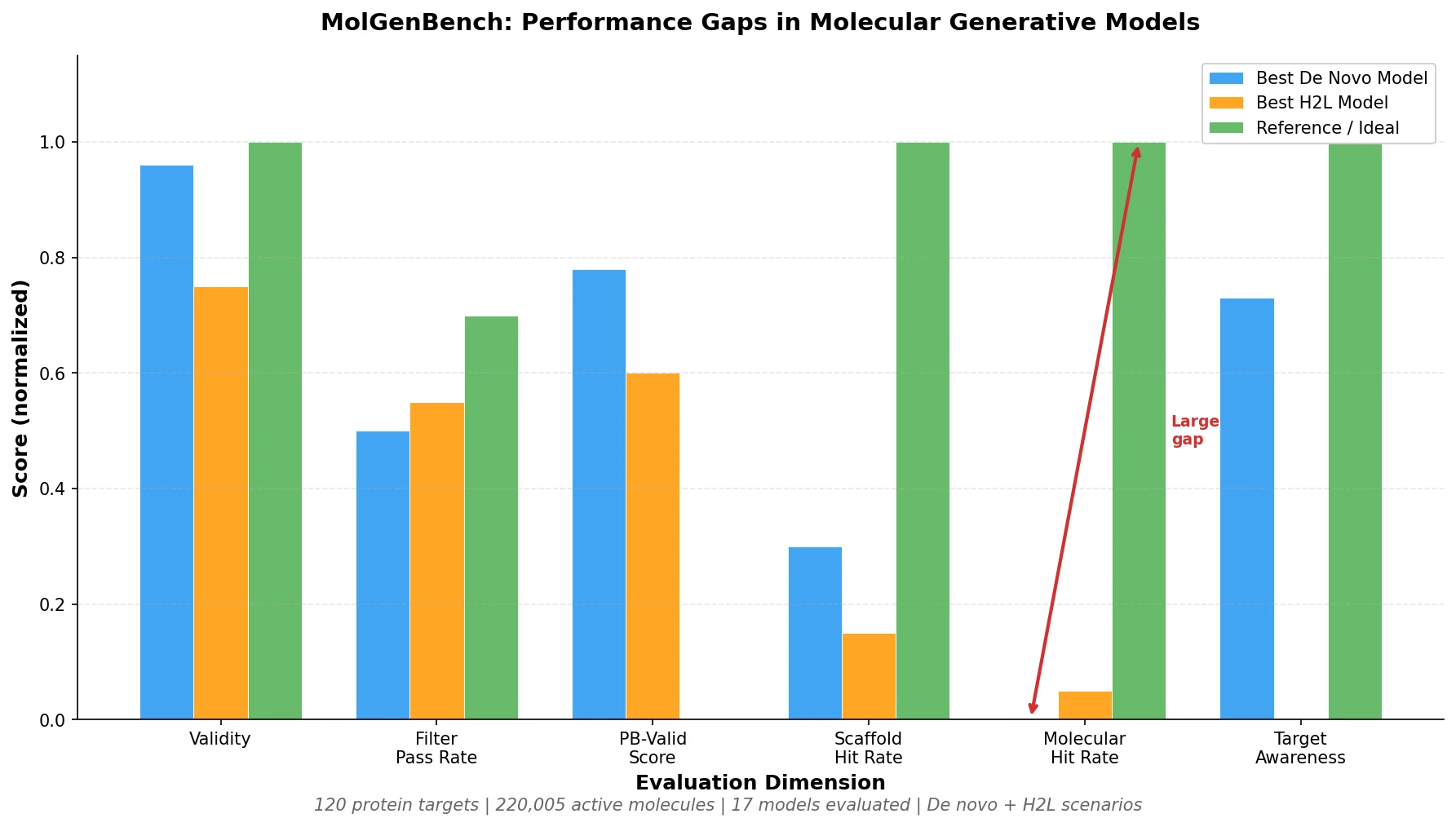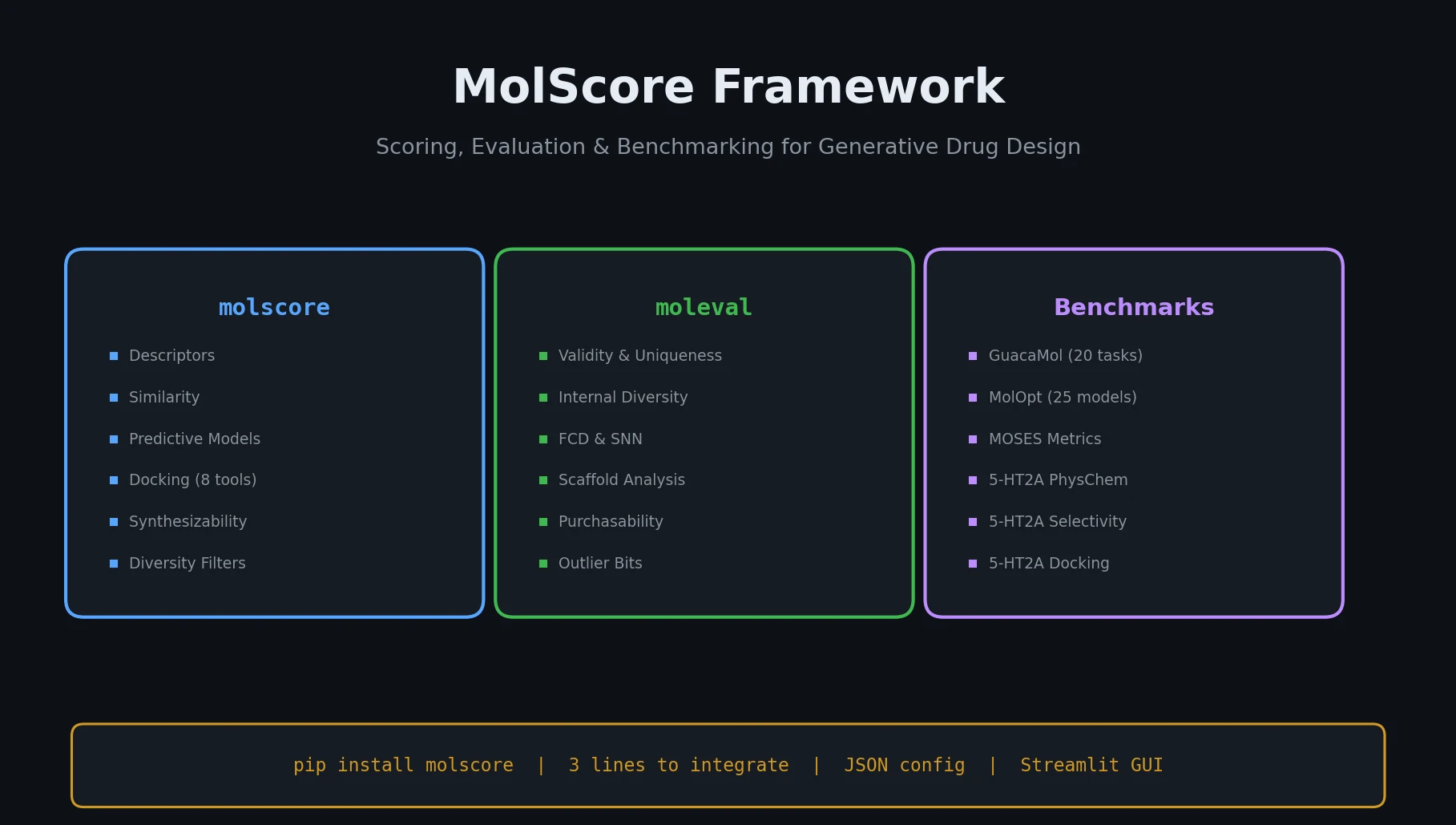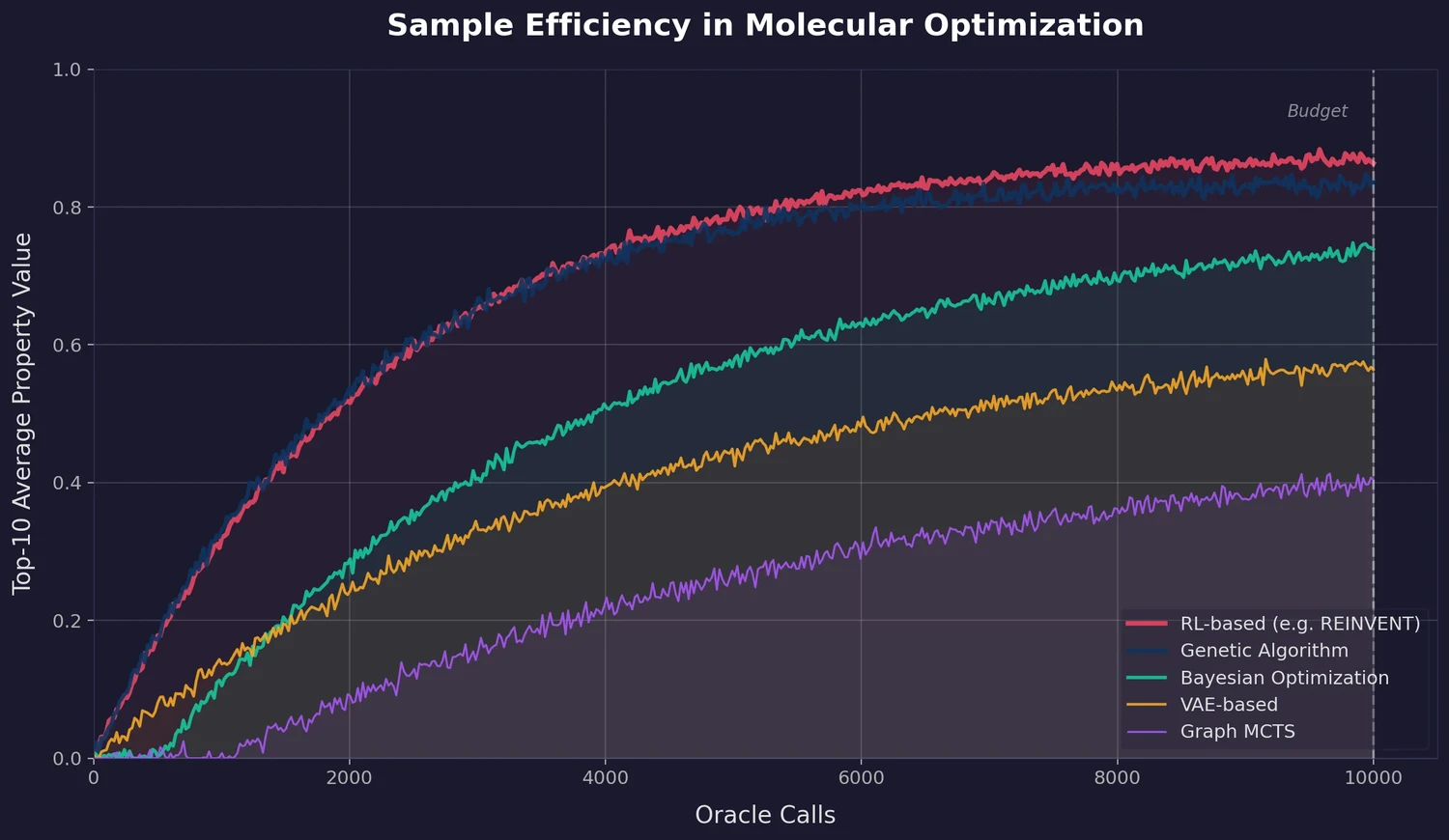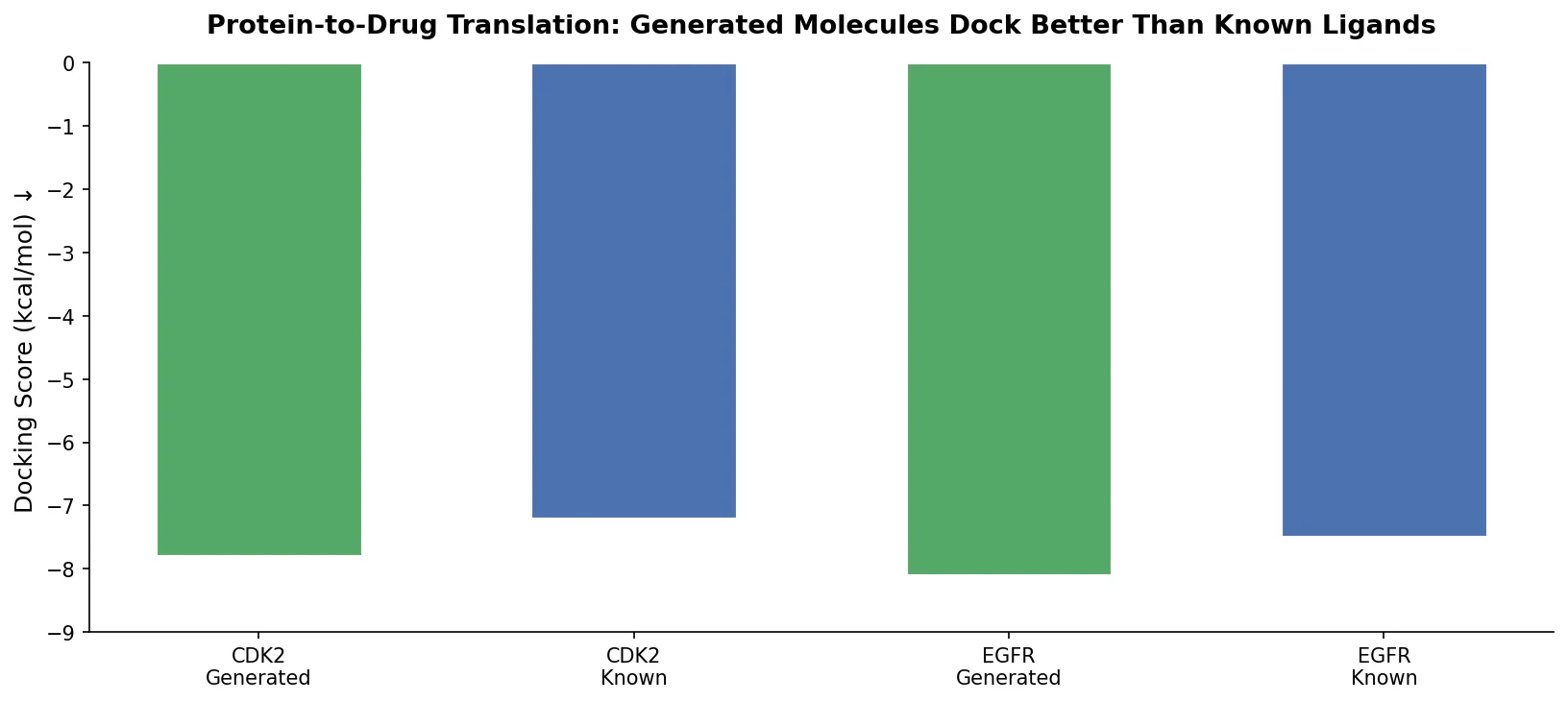
Protein-to-Drug Molecule Translation via Transformer
Applies the Transformer architecture to generate drug-like molecules conditioned on protein amino acid sequences, treating target-specific de novo drug design as a sequence-to-sequence translation problem.