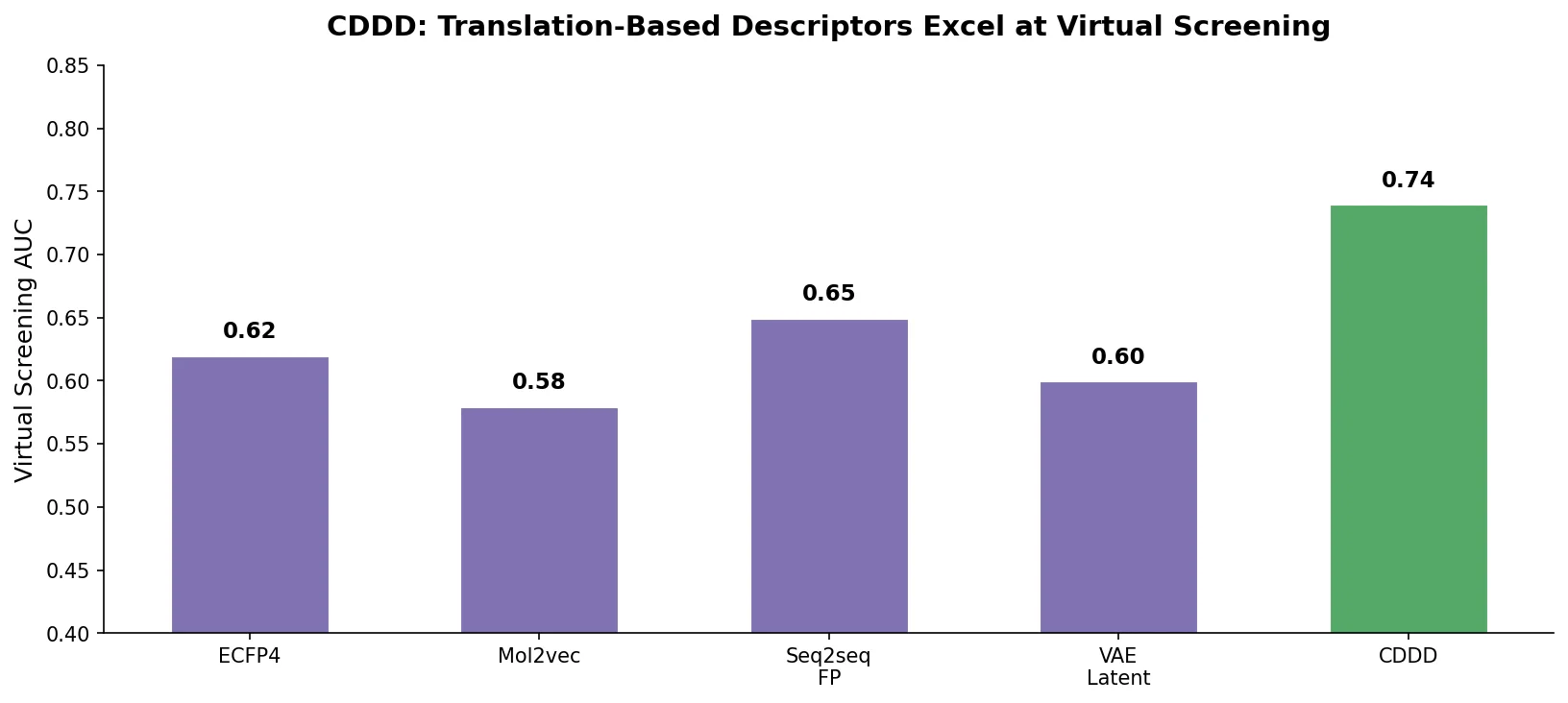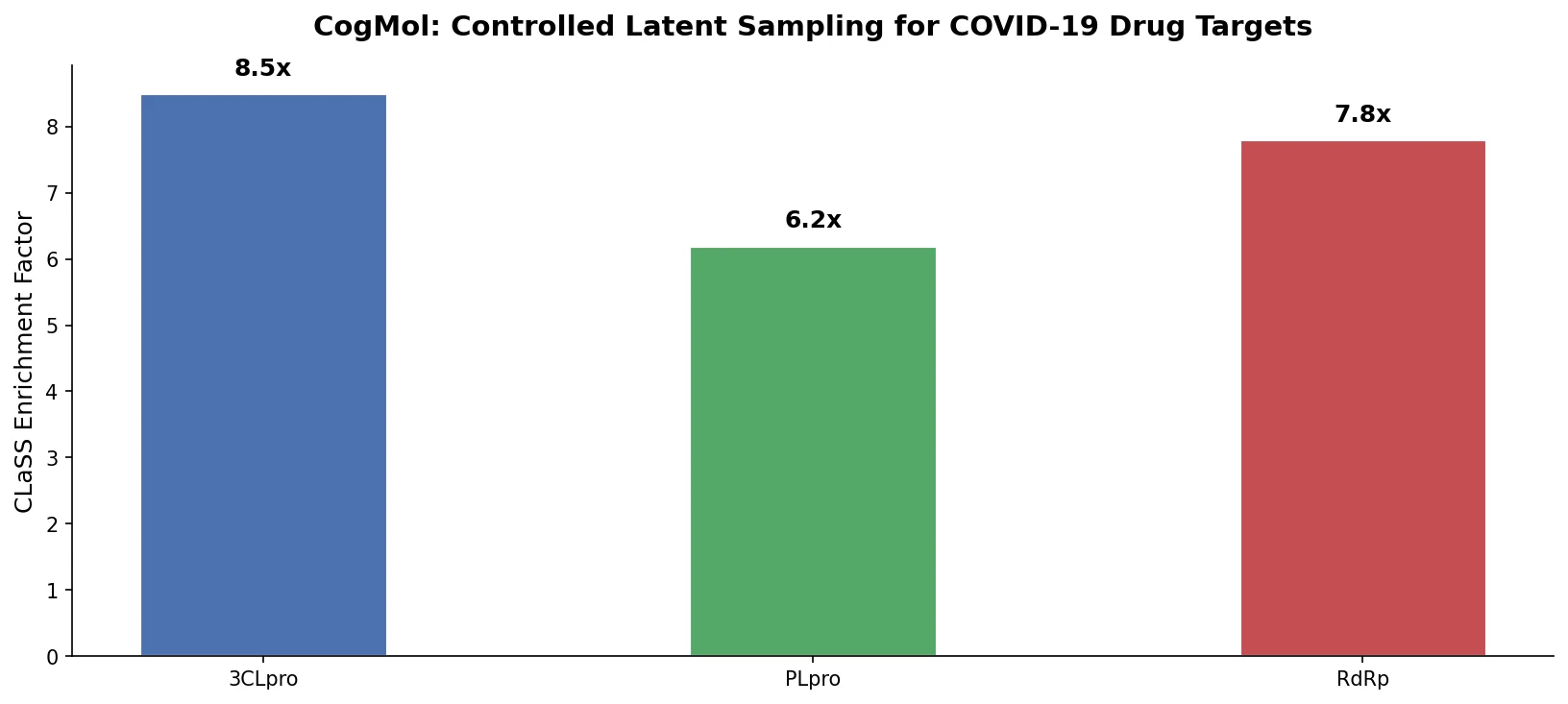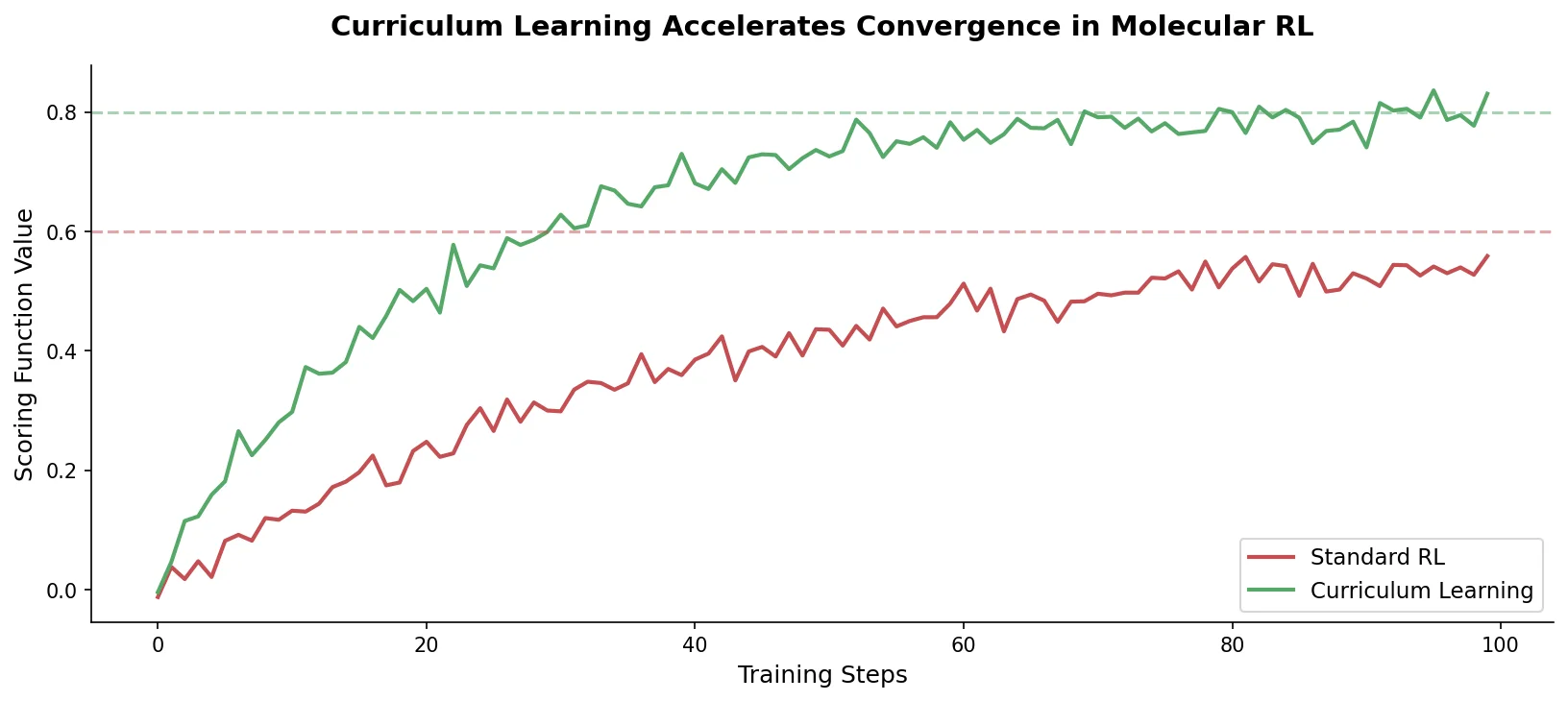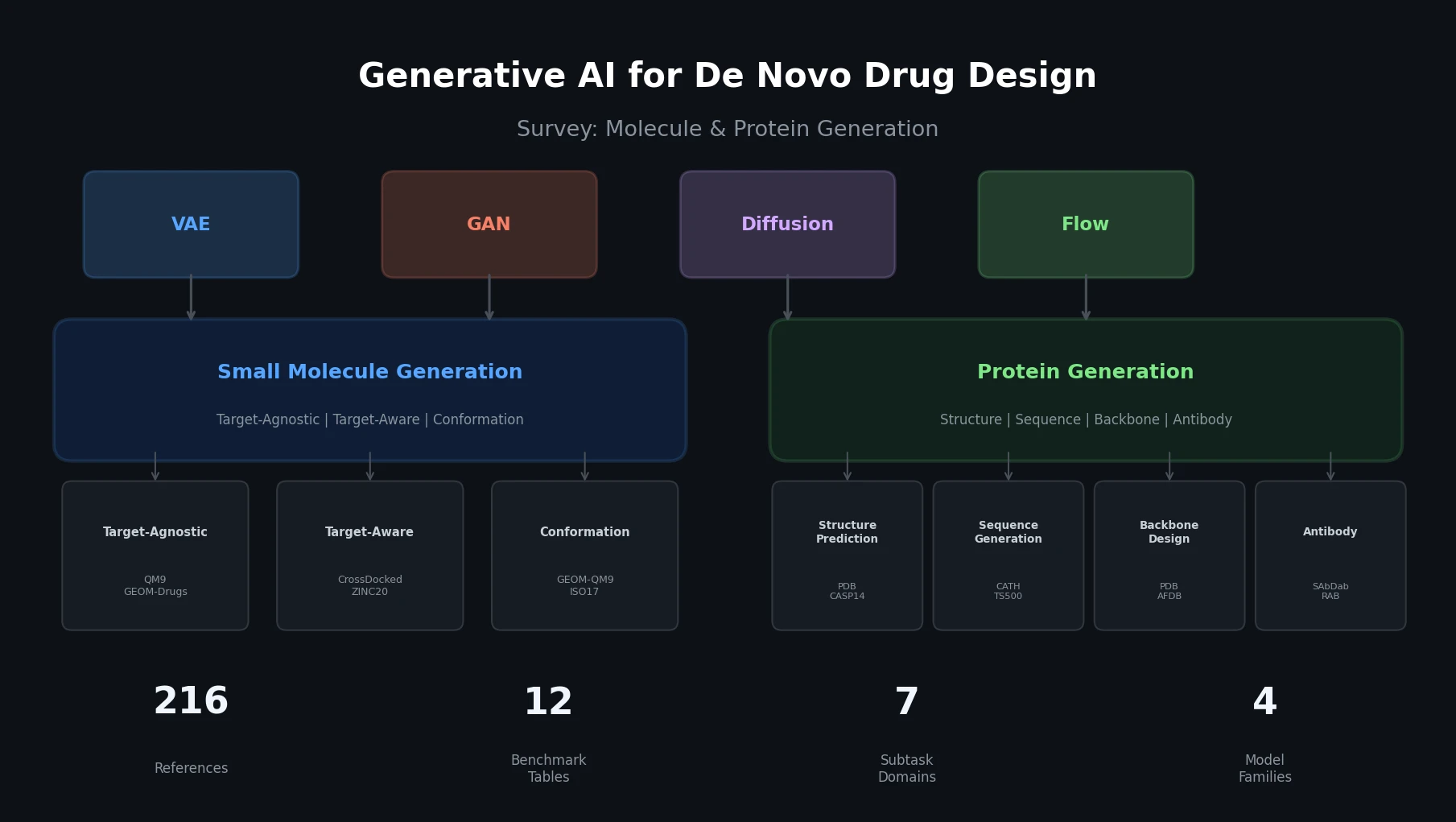
MG-BERT: Graph BERT for Molecular Property Prediction
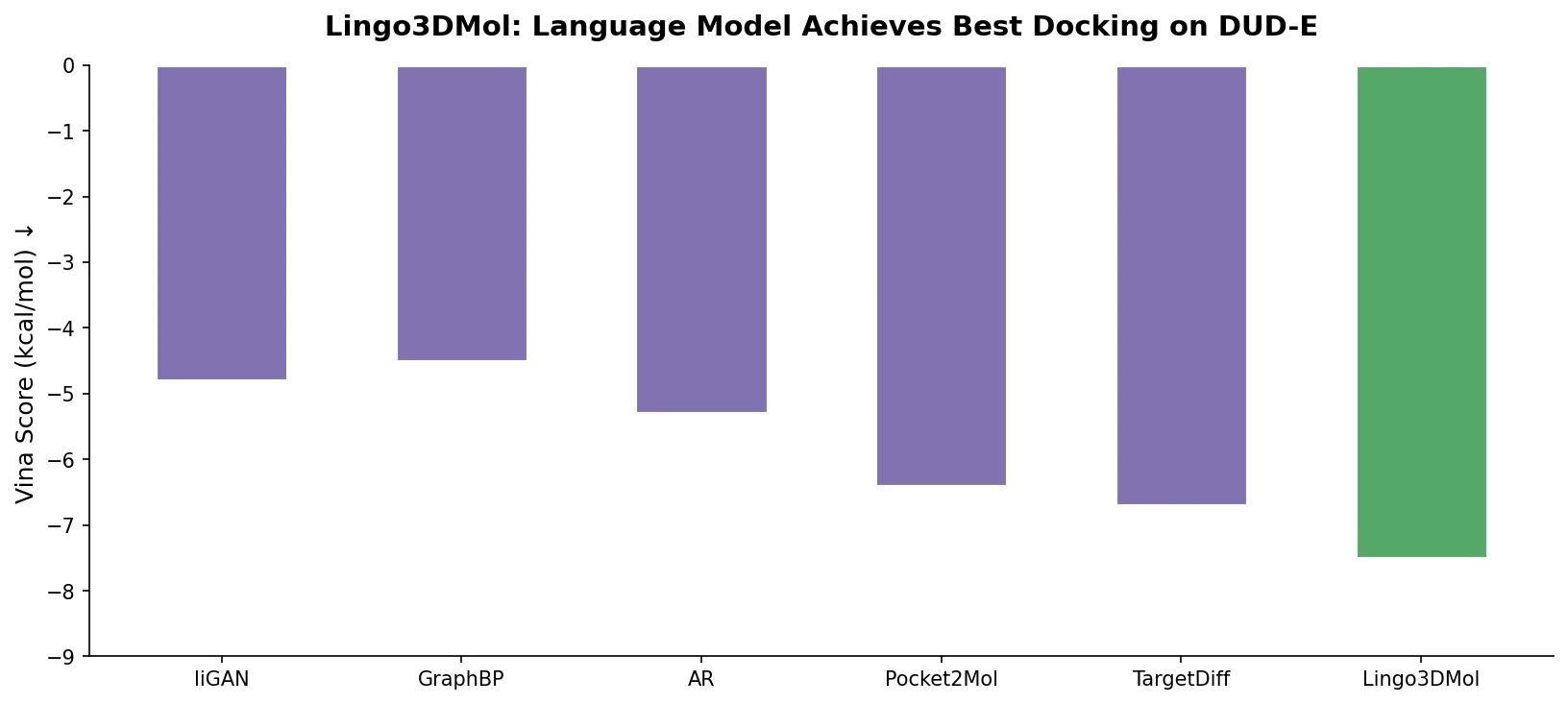
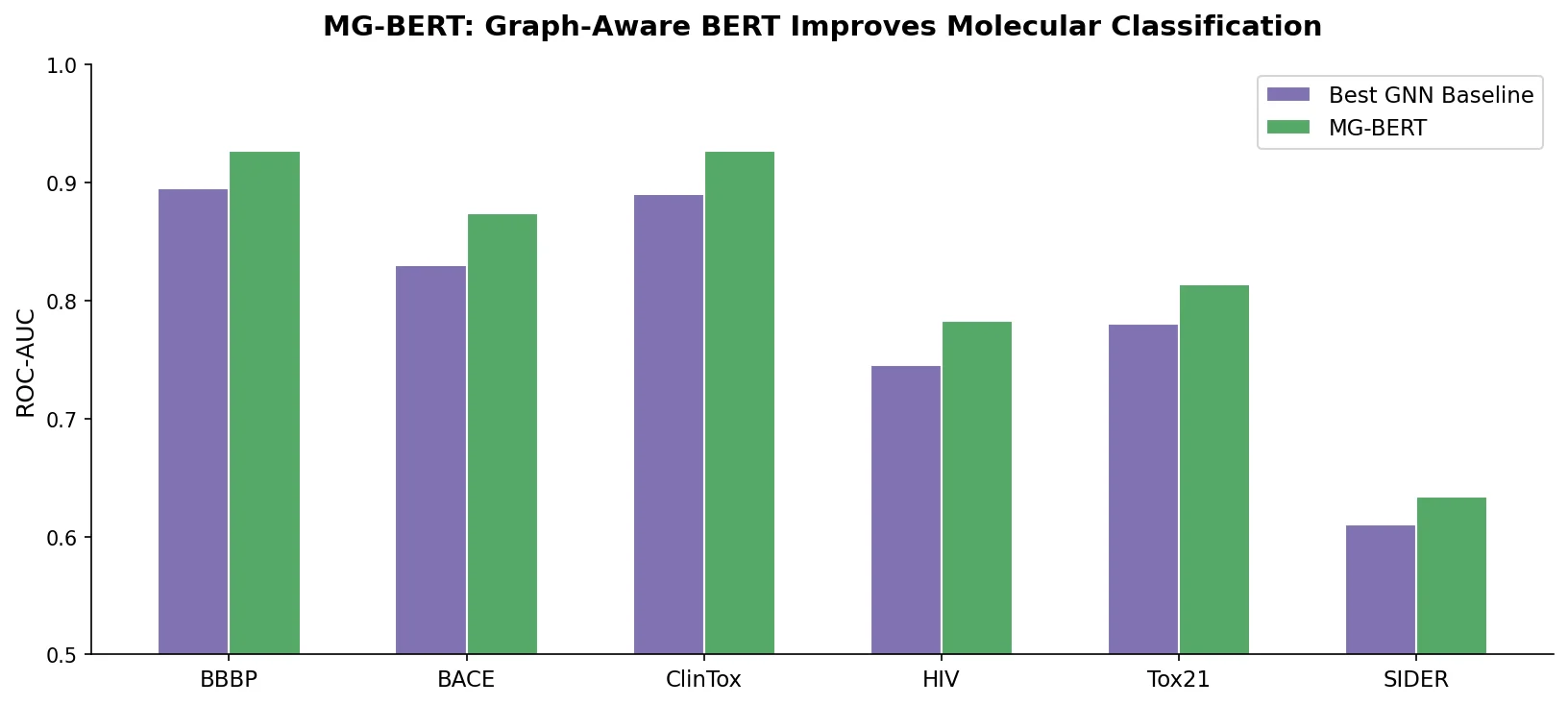
MG-BERT combines GNN-style local attention with BERT’s masked pretraining on molecular graphs, learning context-sensitive atomic representations that improve ADMET property prediction across 11 benchmark datasets.