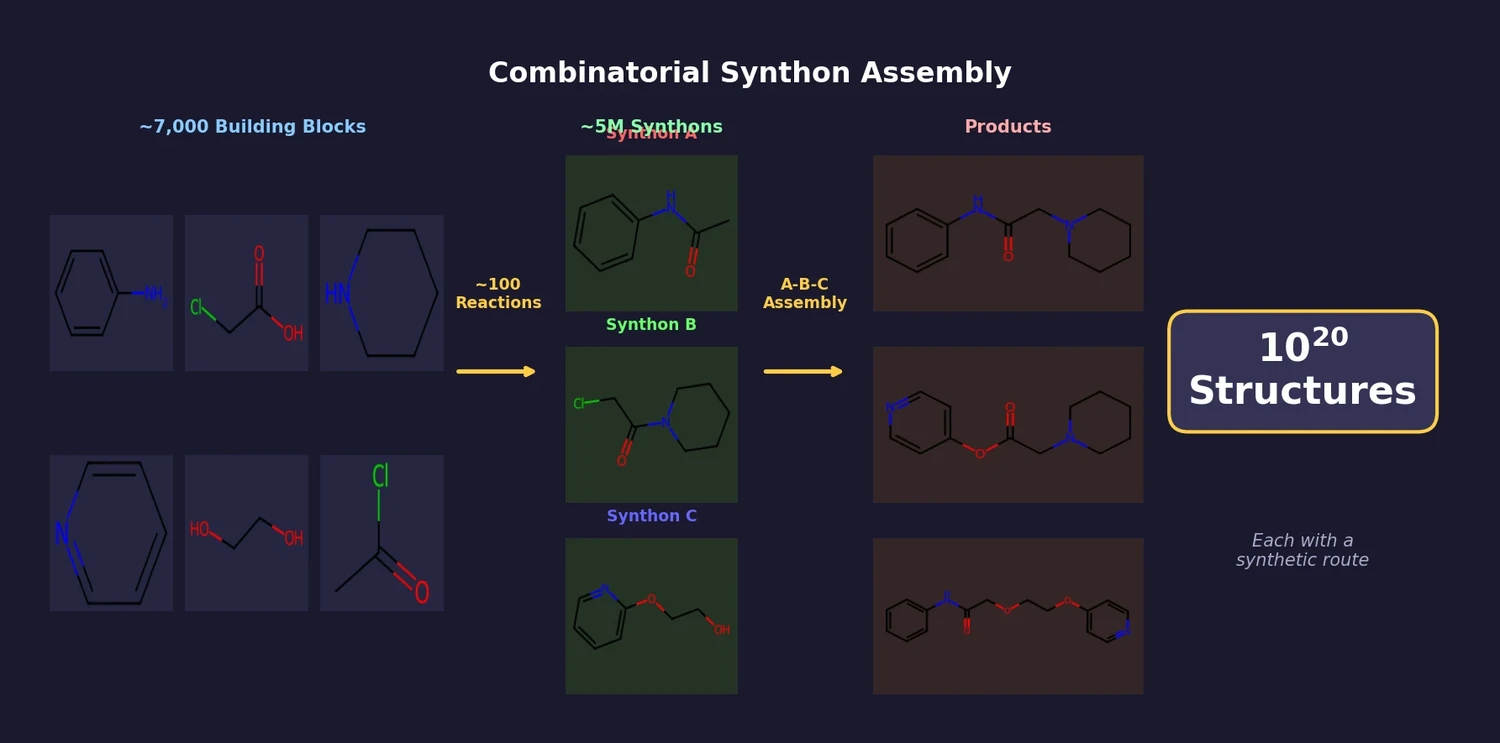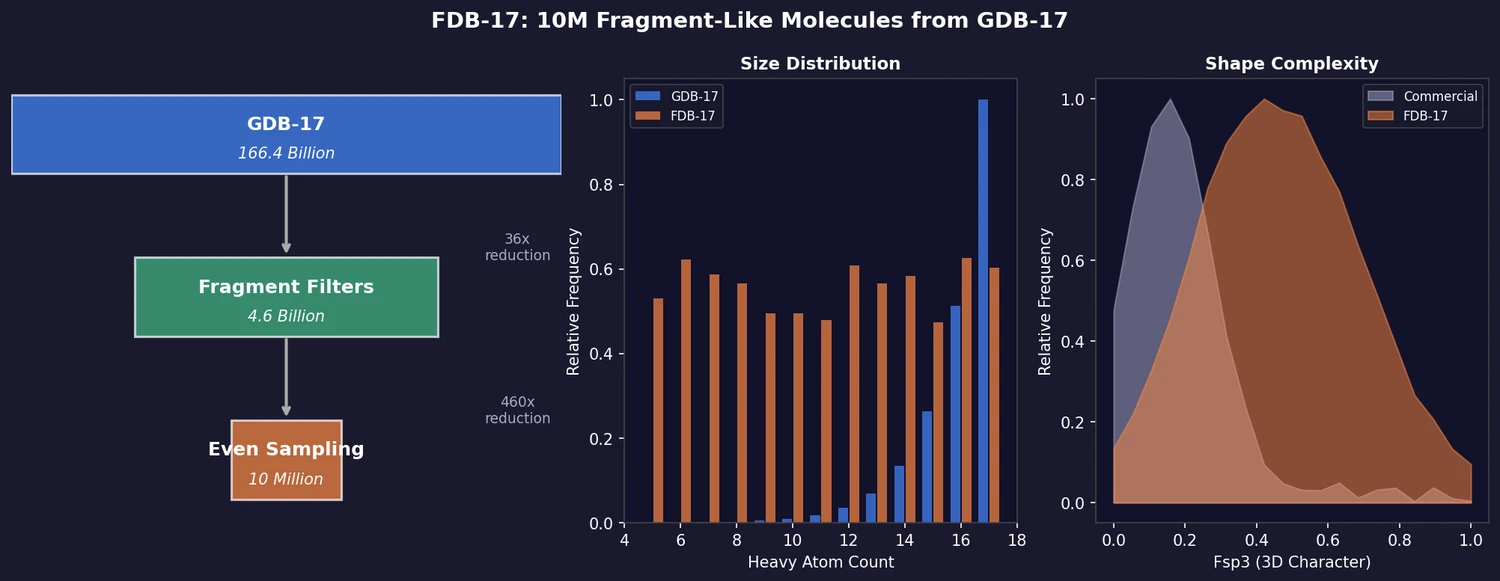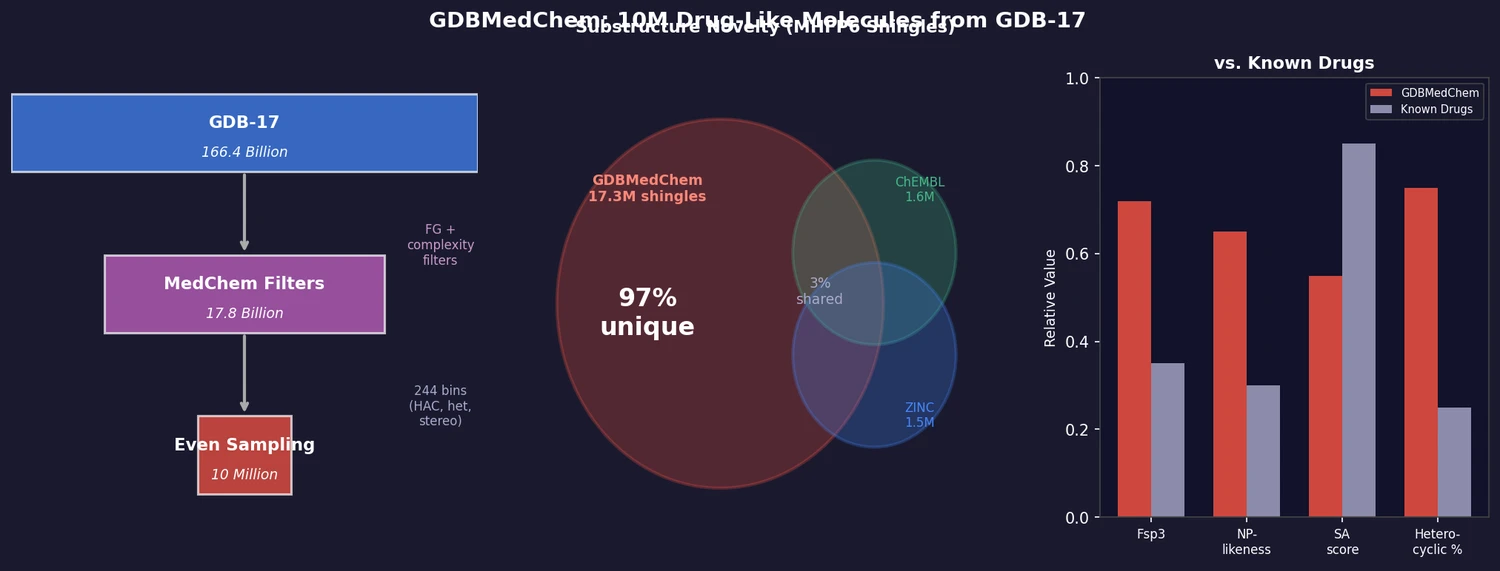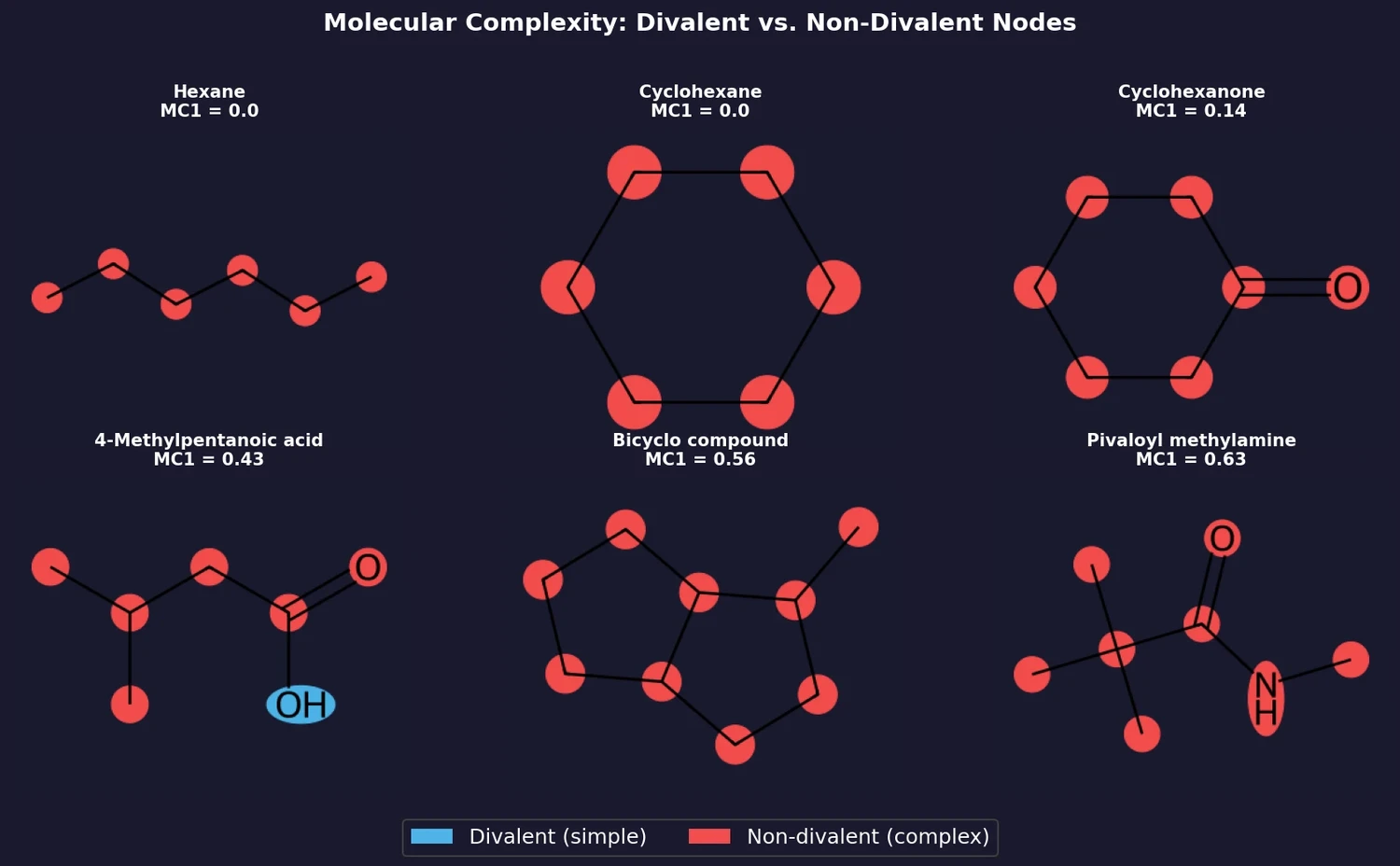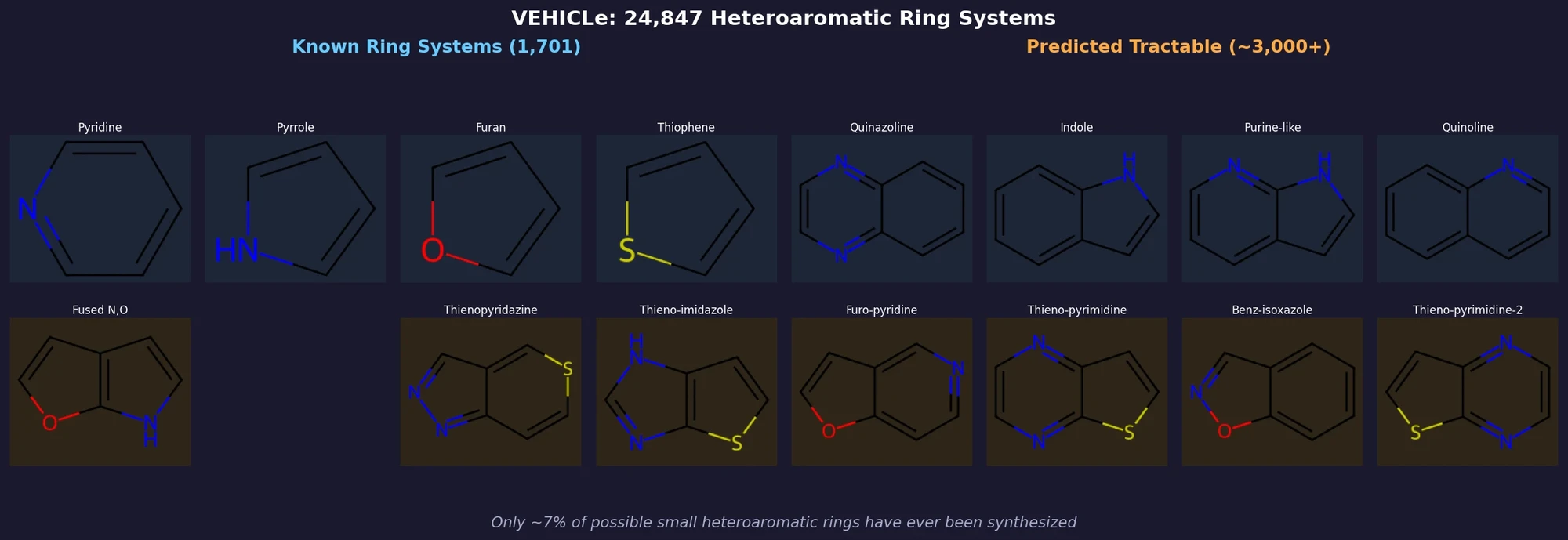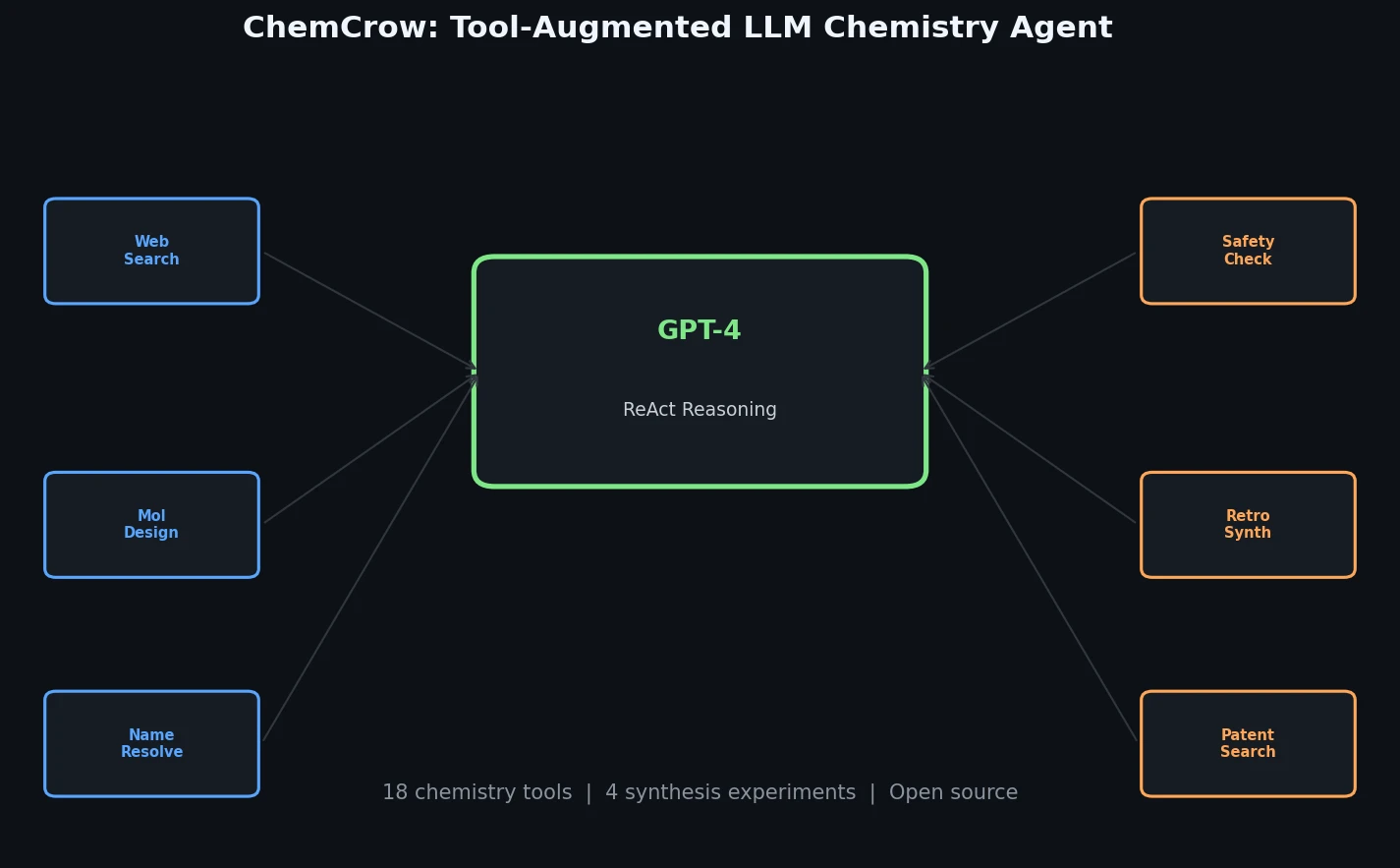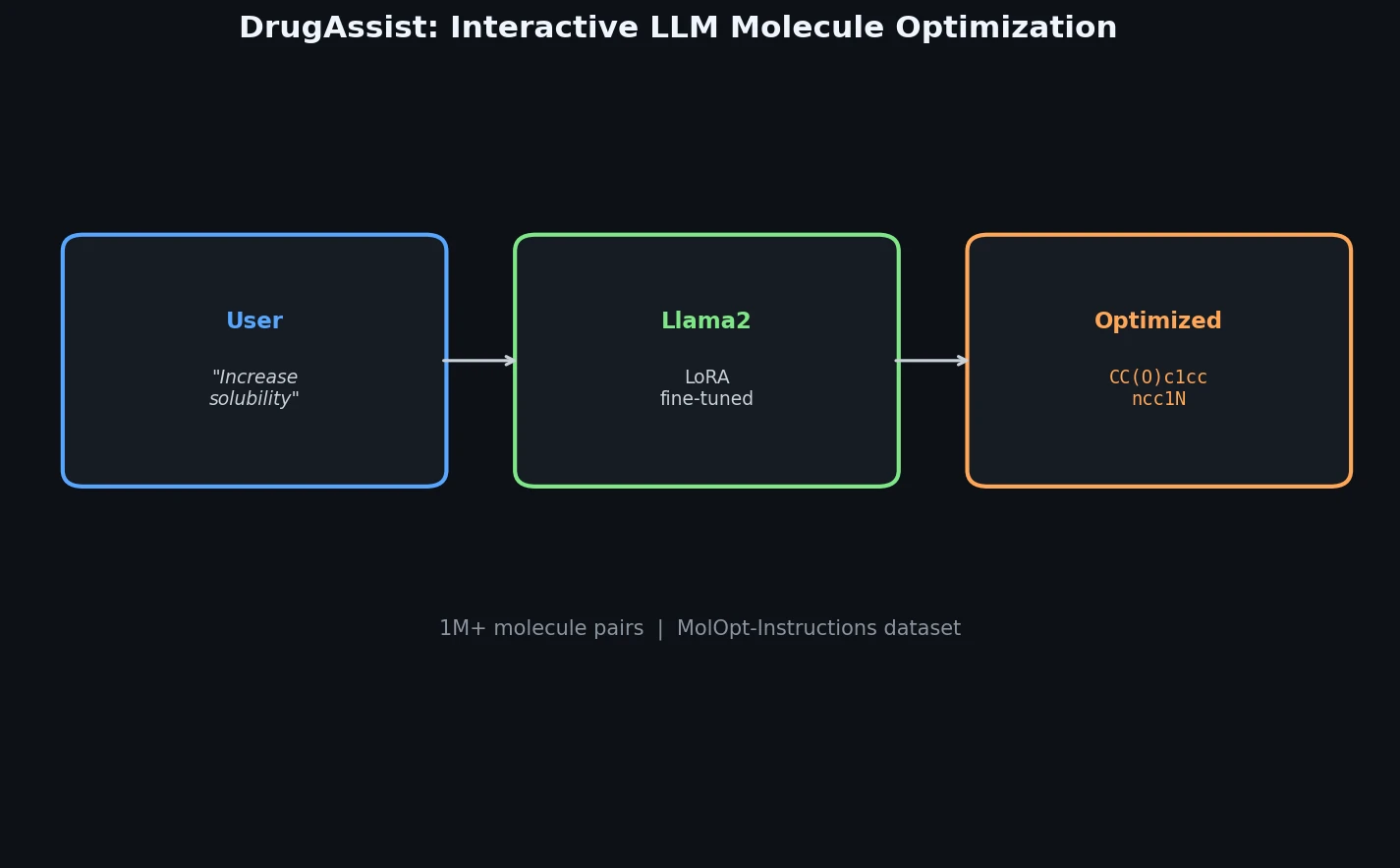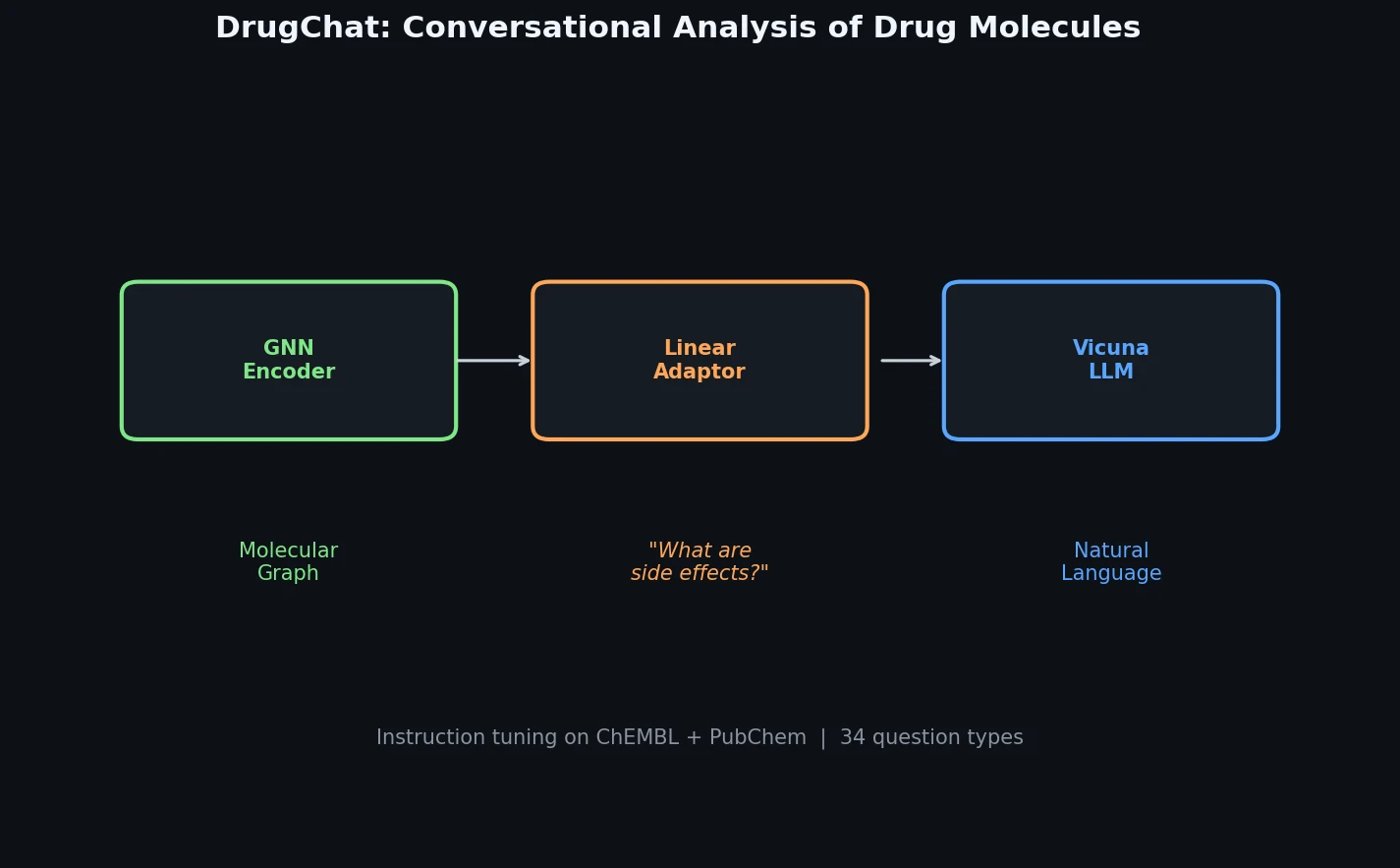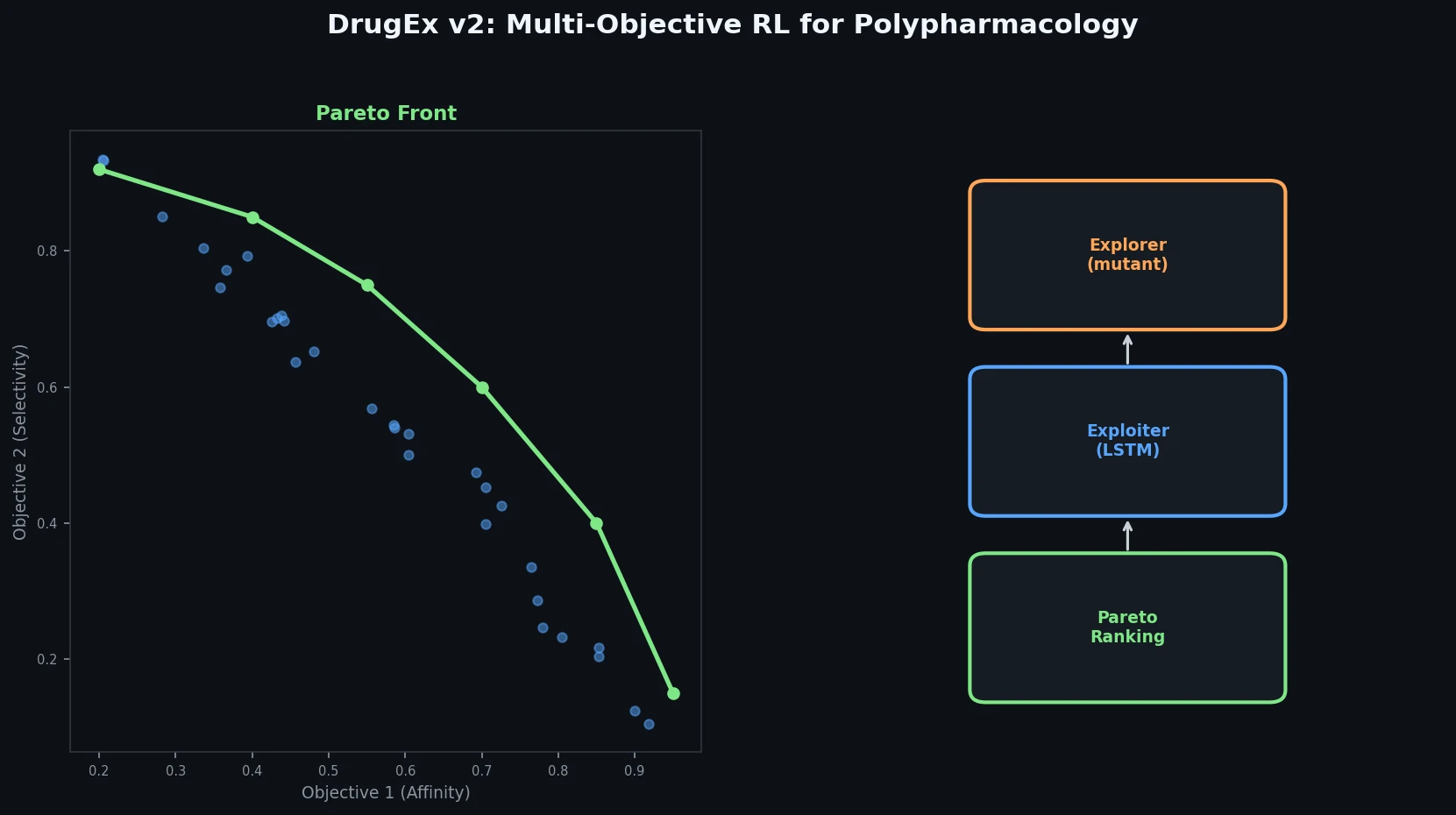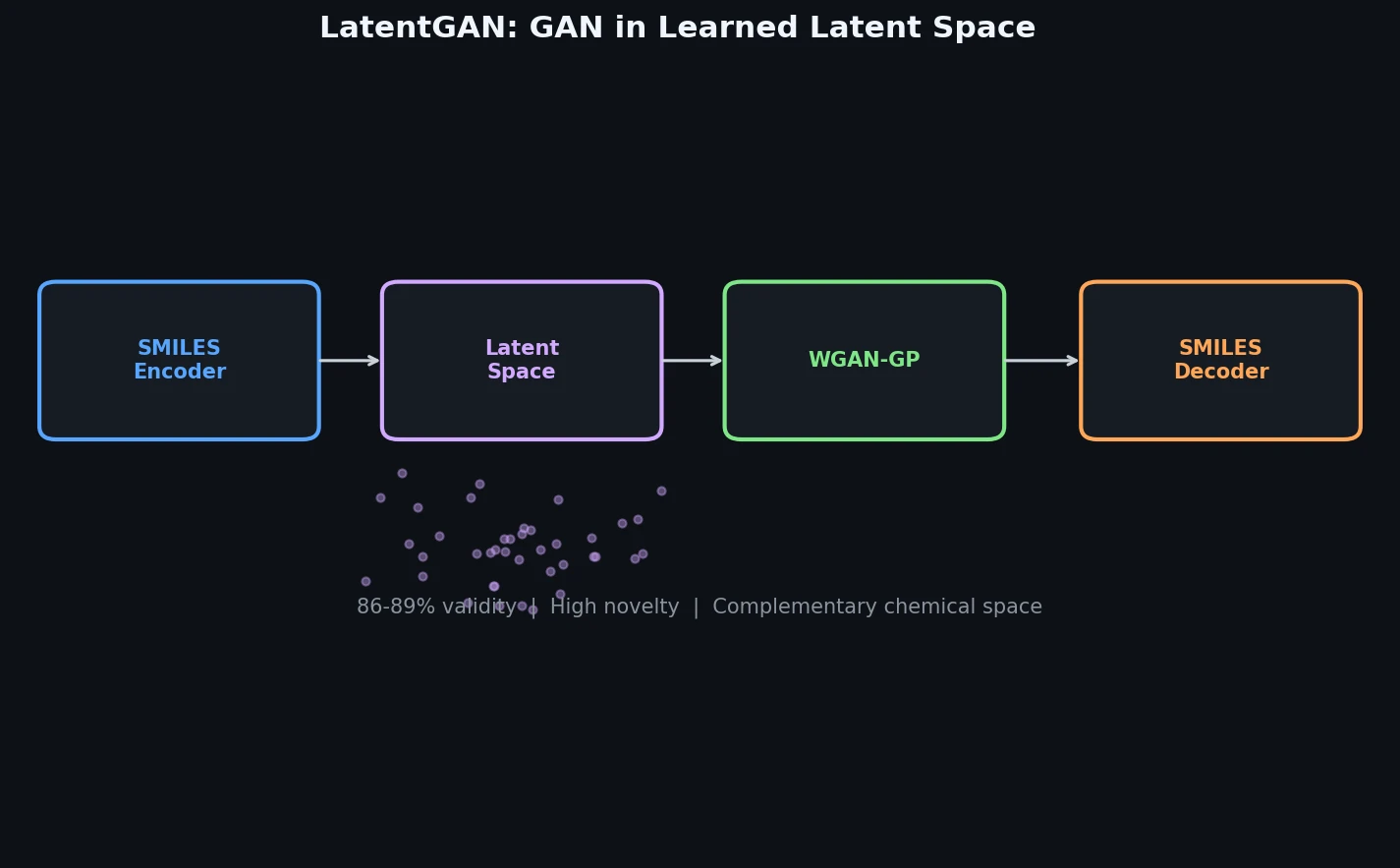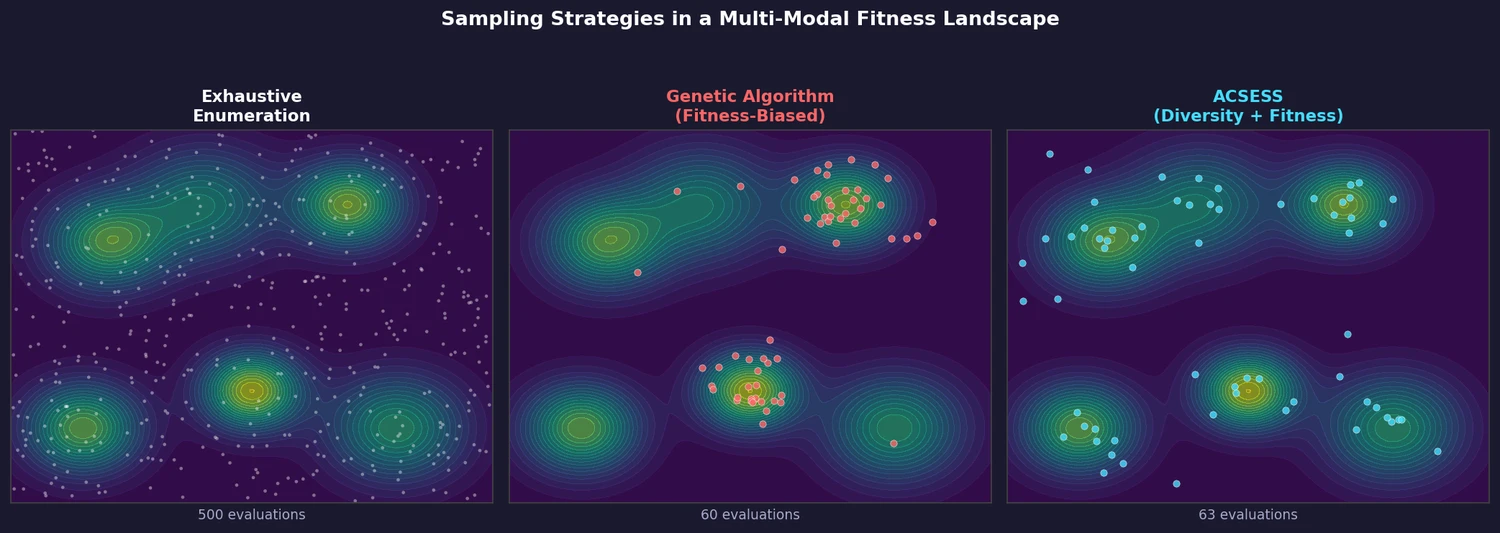
ACSESS: Diverse Optimal Molecules in the SMU
Property-optimizing ACSESS combines diversity-biased sampling with iterative fitness thresholding to discover diverse sets of molecules with favorable properties. Tested on GDB-9 (dipole moment optimization) and NKp fitness landscapes, it outperforms standard genetic algorithms in diversity while matching or exceeding their fitness, using only ~30,000 evaluations to navigate a 300,000-molecule space.