
ChatDrug: Conversational Drug Editing with ChatGPT
ChatDrug is a parameter-free framework that combines ChatGPT with retrieval-augmented domain feedback and iterative conversation to edit drugs across small molecules, peptides, and proteins.
ChatDrug is a parameter-free framework that combines ChatGPT with retrieval-augmented domain feedback and iterative conversation to edit drugs across small molecules, peptides, and proteins.
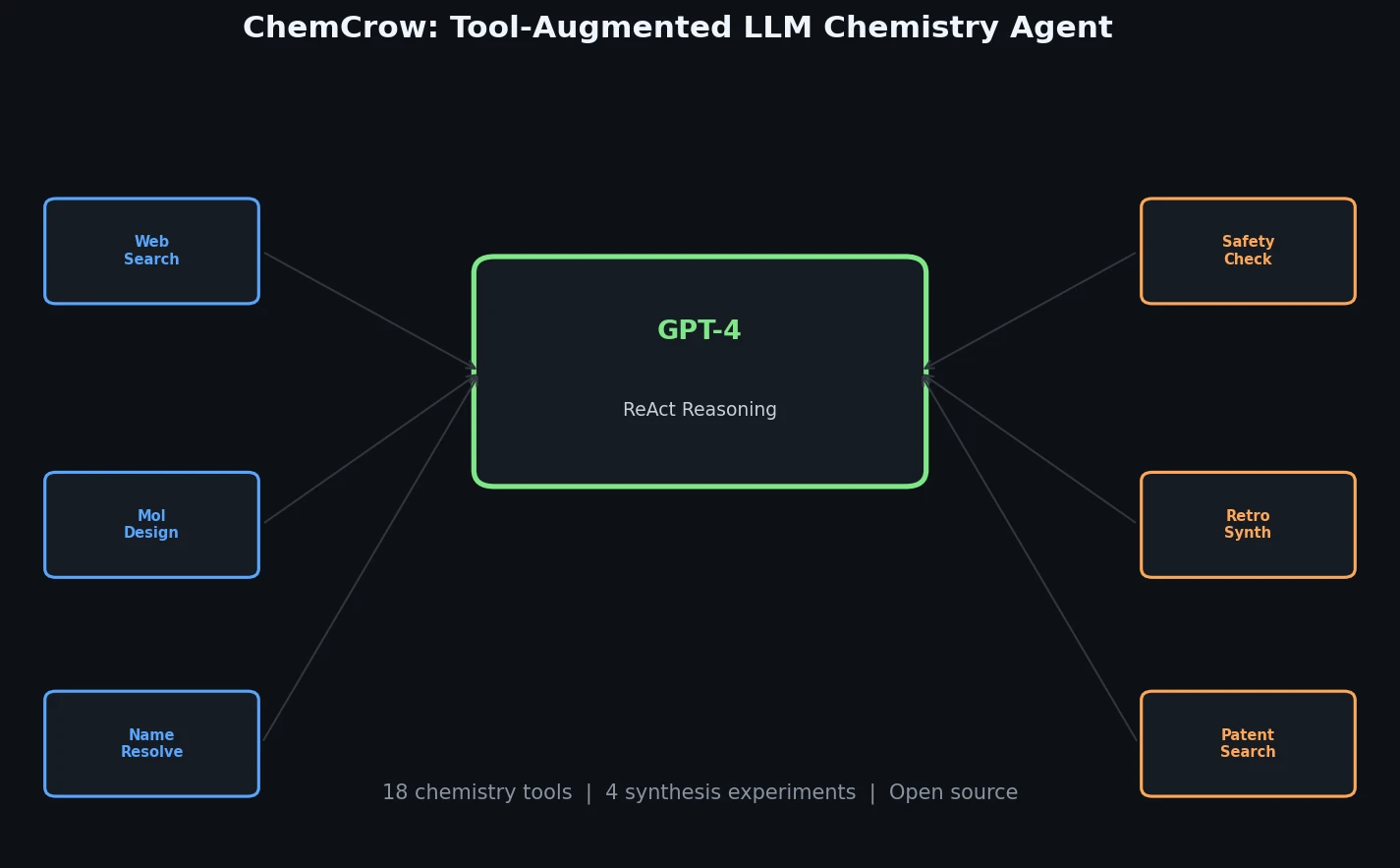
ChemCrow augments GPT-4 with 18 chemistry tools to autonomously plan and execute syntheses, discover novel chromophores, and solve diverse chemical reasoning tasks.
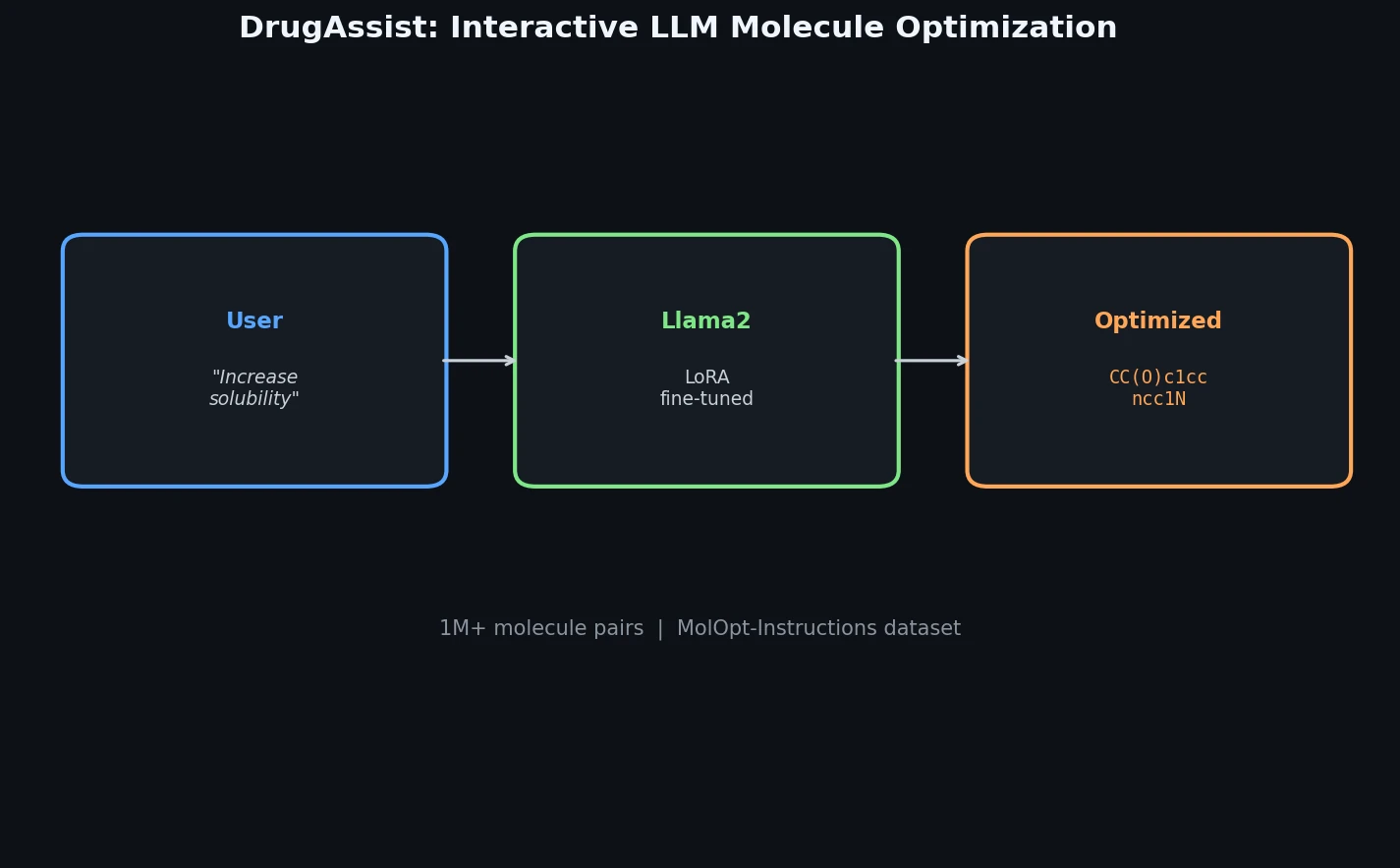
DrugAssist fine-tunes Llama2-7B-Chat on over one million molecule pairs for interactive, dialogue-based molecule optimization across six molecular properties.
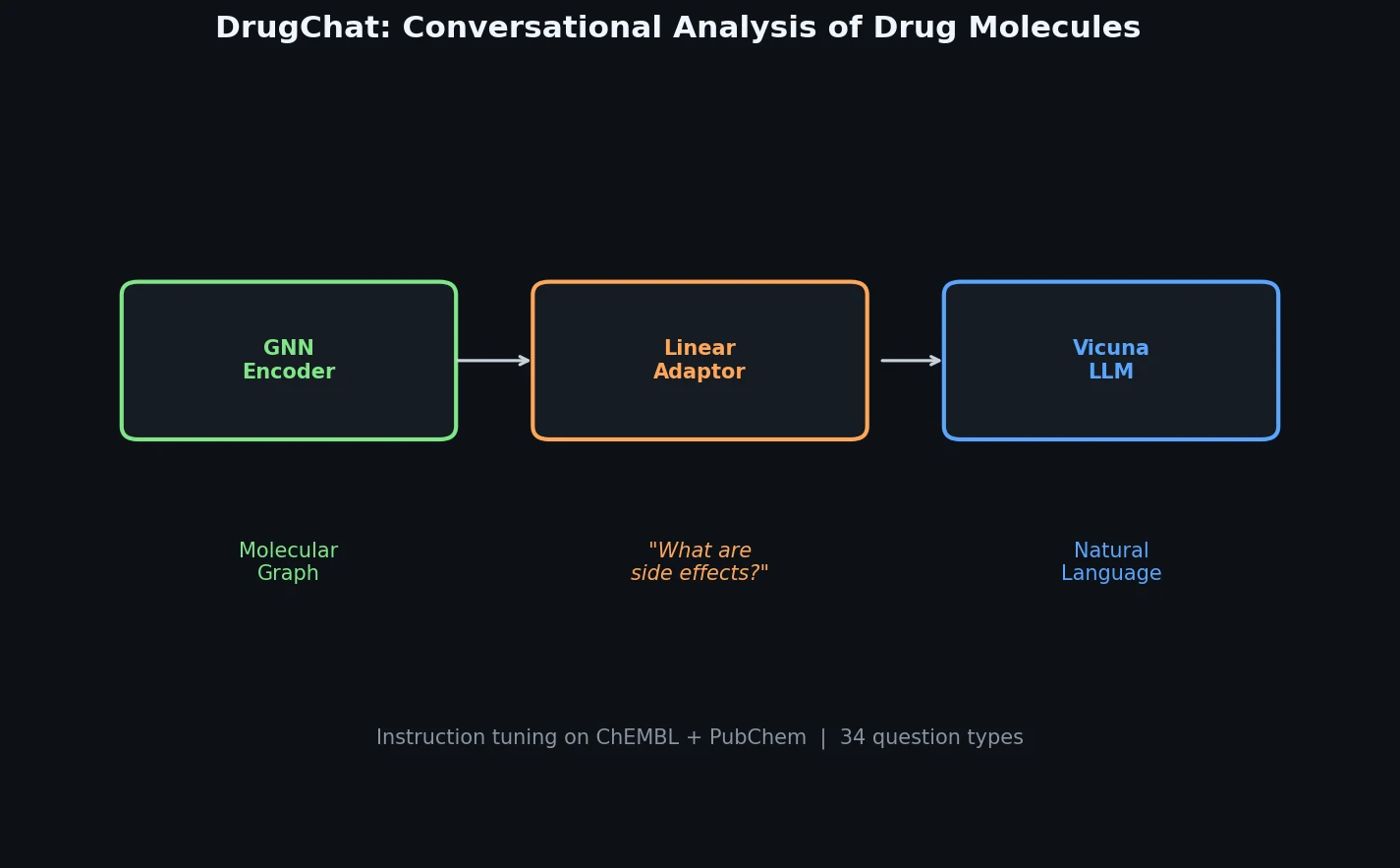
DrugChat is a prototype system that bridges molecular graph neural networks with large language models for interactive, multi-turn question answering about drug compounds. It trains only a lightweight linear adaptor between a frozen GNN encoder and Vicuna-13B using 143K curated QA pairs from ChEMBL and PubChem.
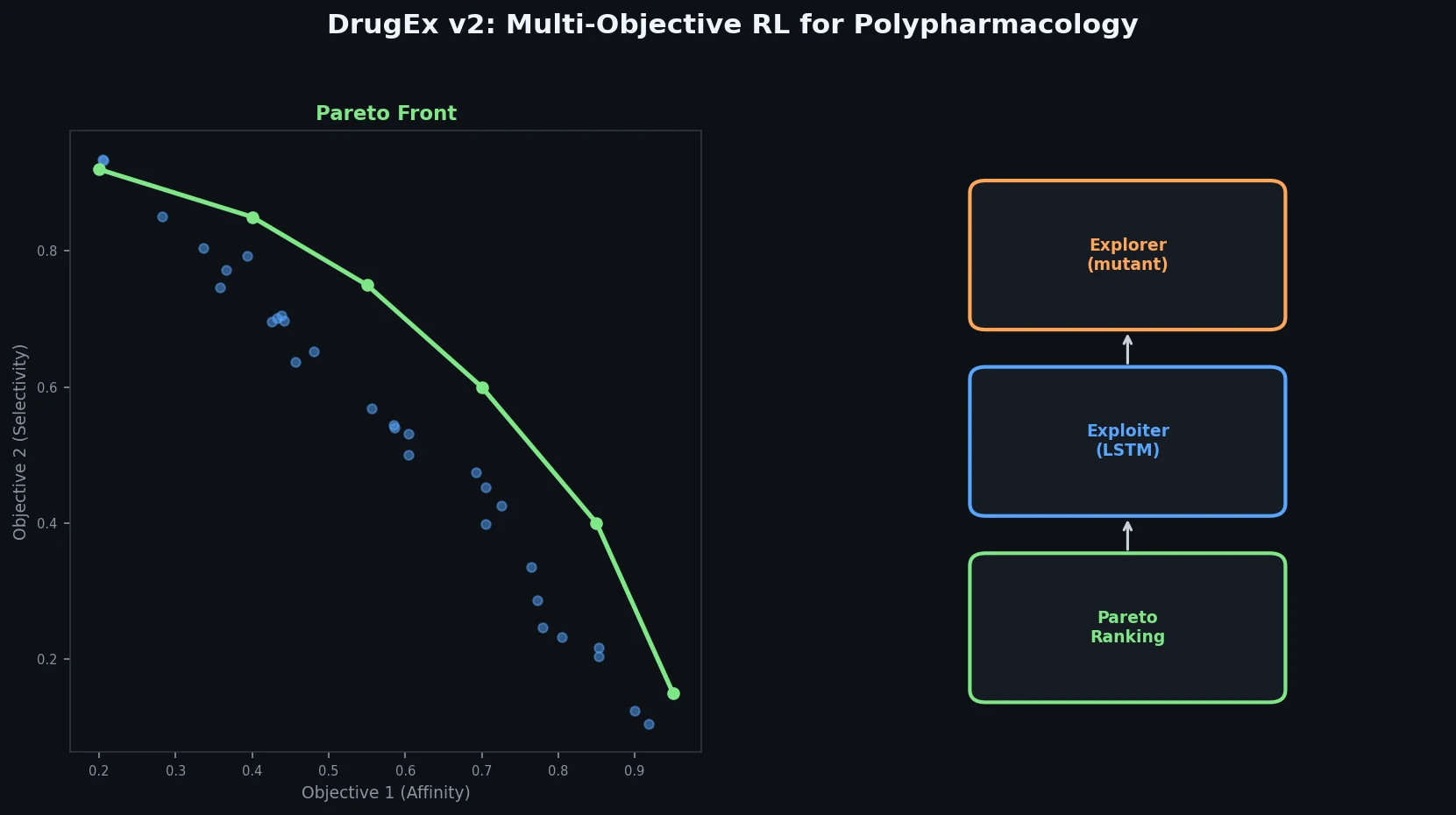
DrugEx v2 introduces Pareto-based multi-objective optimization and evolutionary exploration strategies into an RNN reinforcement learning framework for de novo drug design toward multiple protein targets.
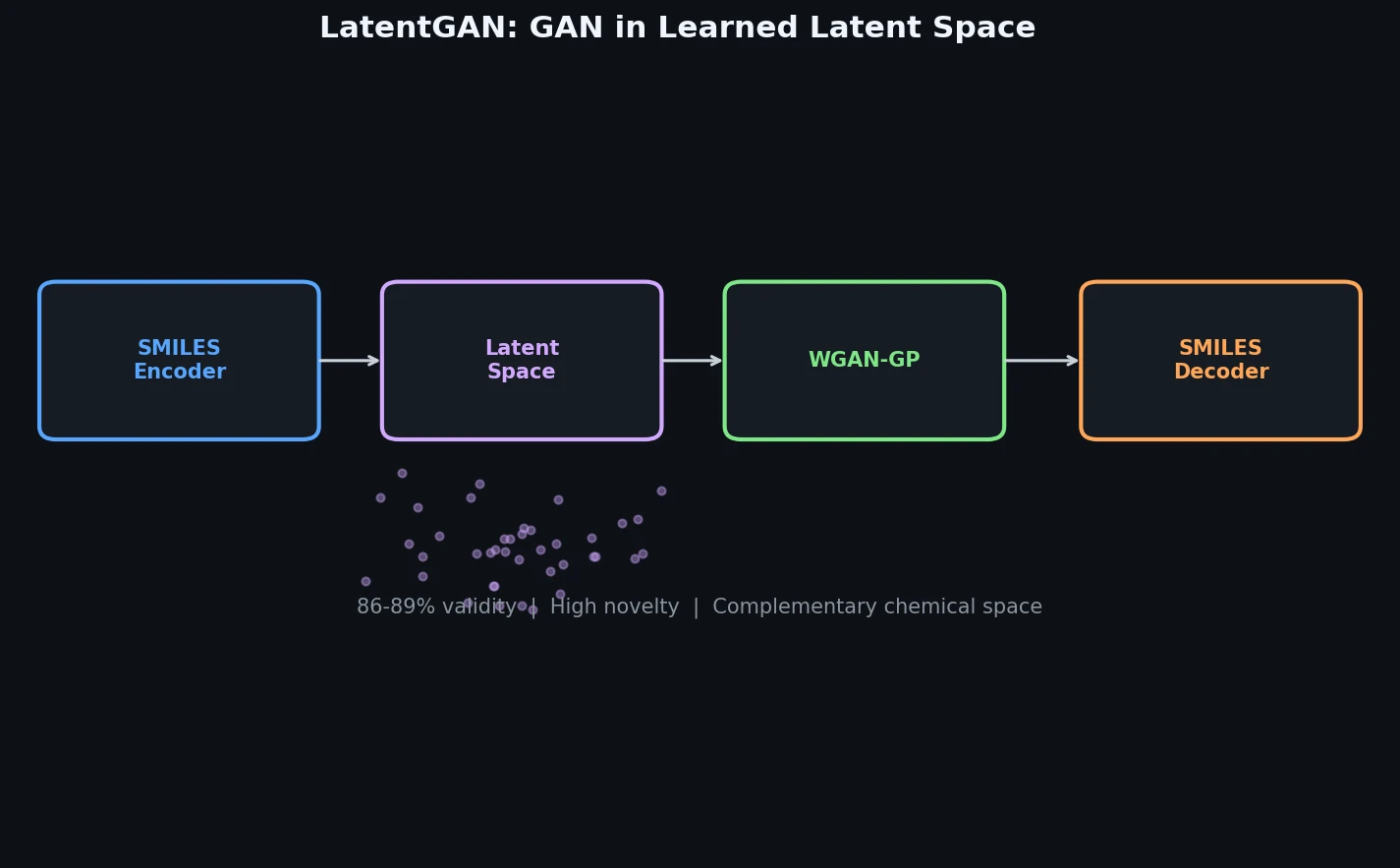
LatentGAN decouples molecular generation from SMILES syntax by training a Wasserstein GAN on latent vectors from a pretrained heteroencoder, enabling de novo design of drug-like and target-biased compounds.
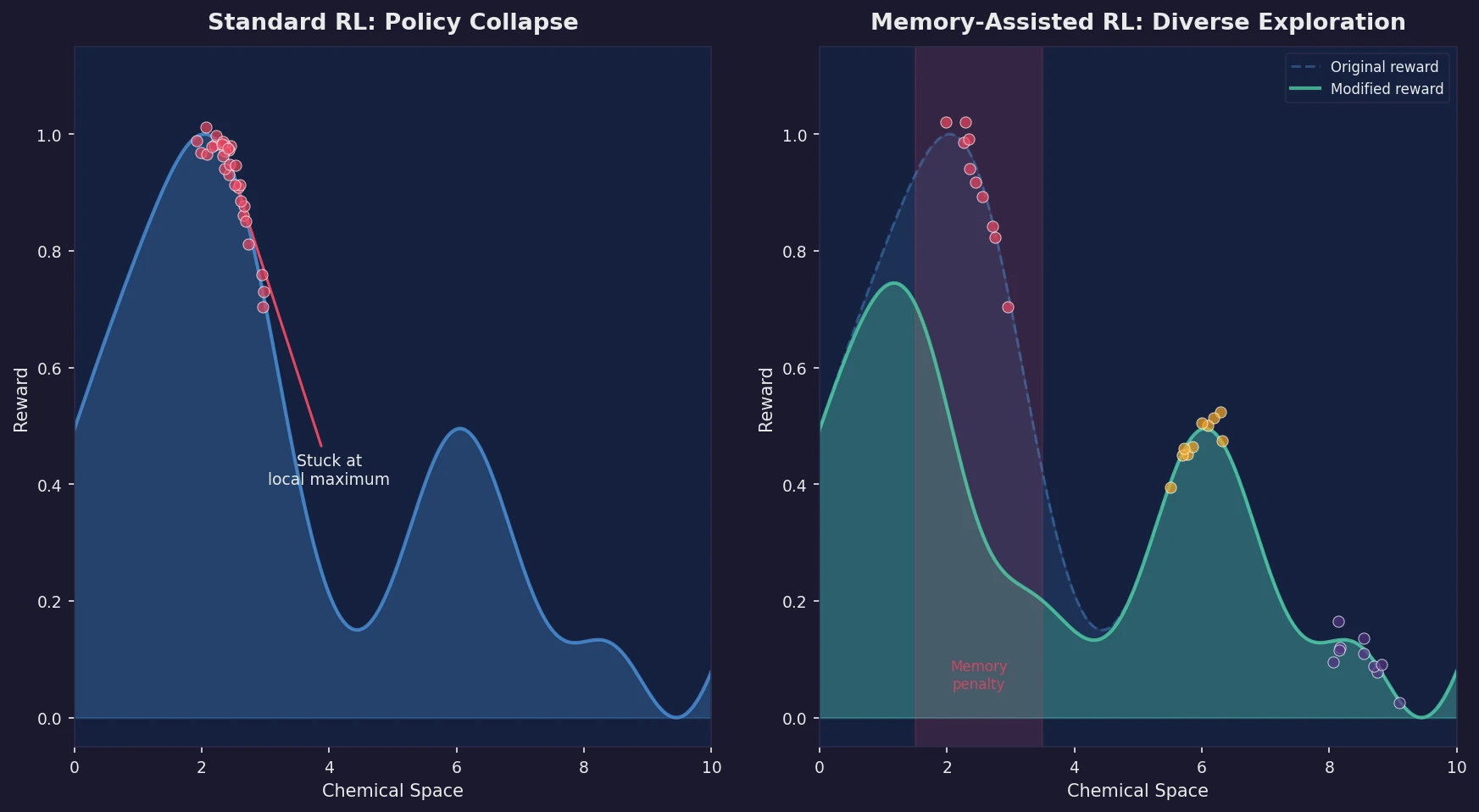
Introduces a memory unit that modifies the RL reward function to penalize previously explored chemical scaffolds, substantially increasing the diversity of generated molecules while maintaining relevance to known active ligands.
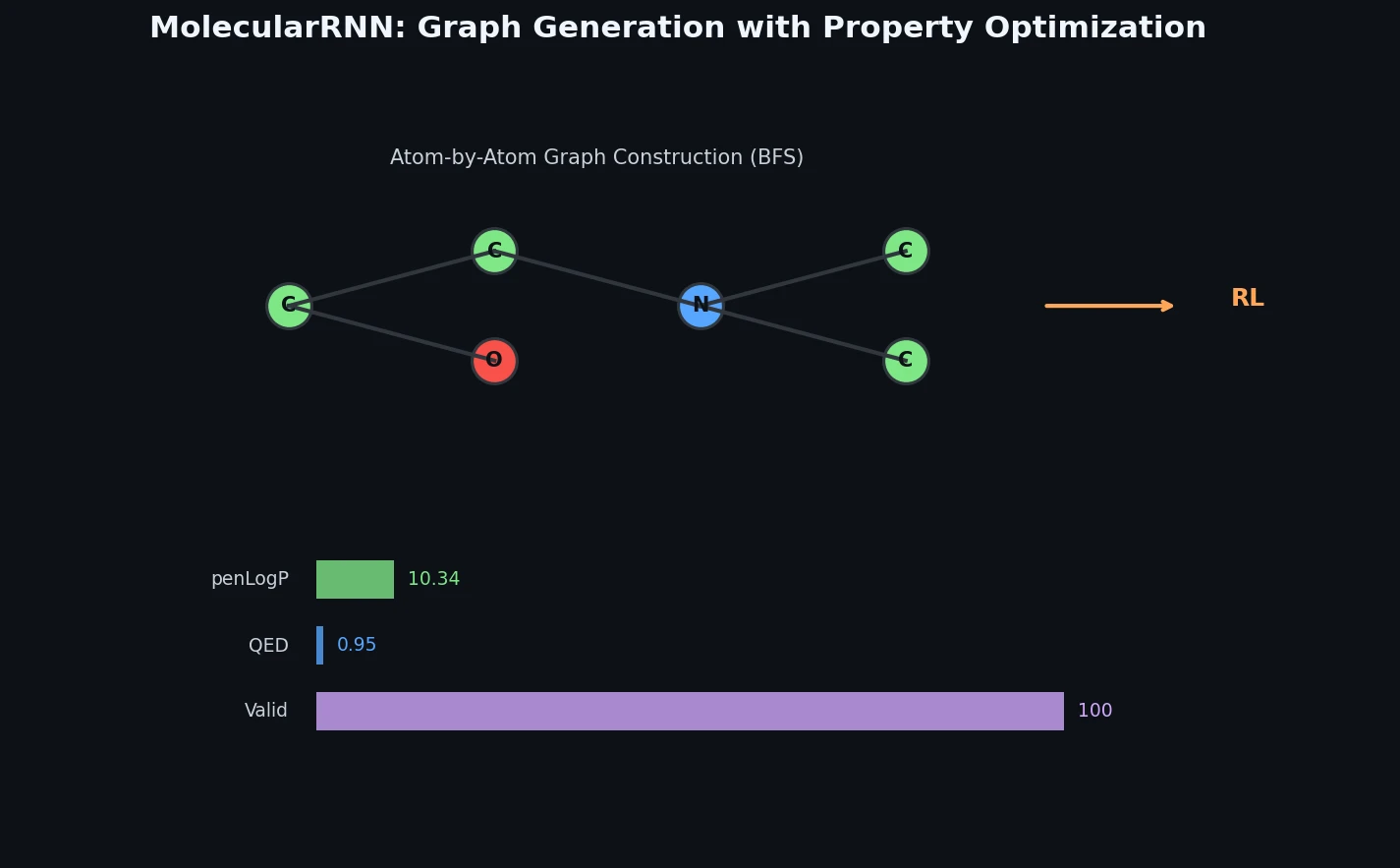
Proposes MolecularRNN, a graph recurrent model that generates molecular graphs atom-by-atom with 100% validity via valency-based rejection sampling, then shifts property distributions using policy gradient reinforcement learning.
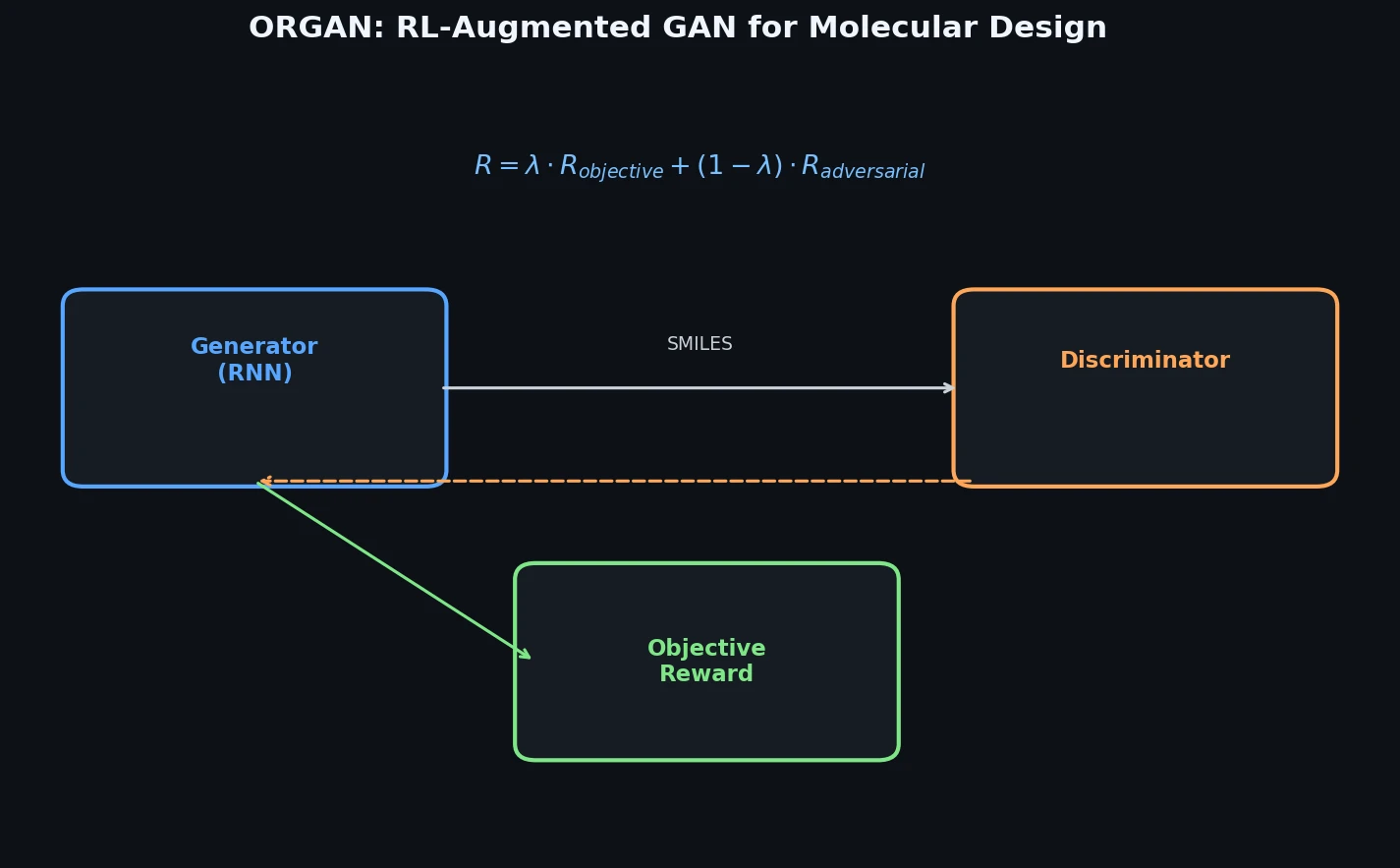
Proposes ORGAN, a framework that extends SeqGAN with domain-specific reward functions via reinforcement learning, enabling tunable generation of molecules optimized for druglikeness, solubility, and synthesizability while maintaining sample diversity.
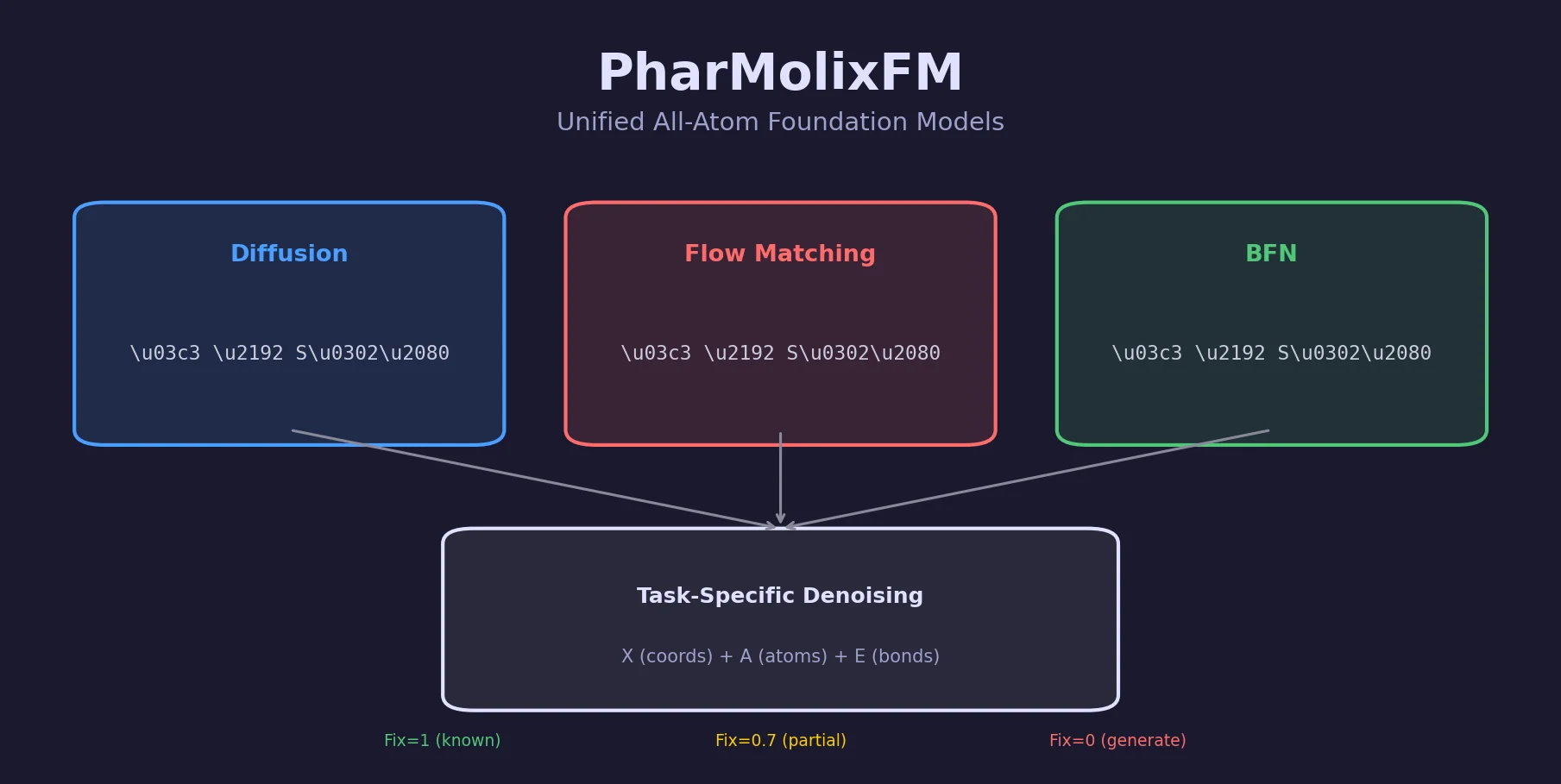
PharMolixFM proposes a unified framework for all-atom foundation models using three multi-modal generative approaches (diffusion, flow matching, BFN) and demonstrates competitive docking accuracy with fast inference.
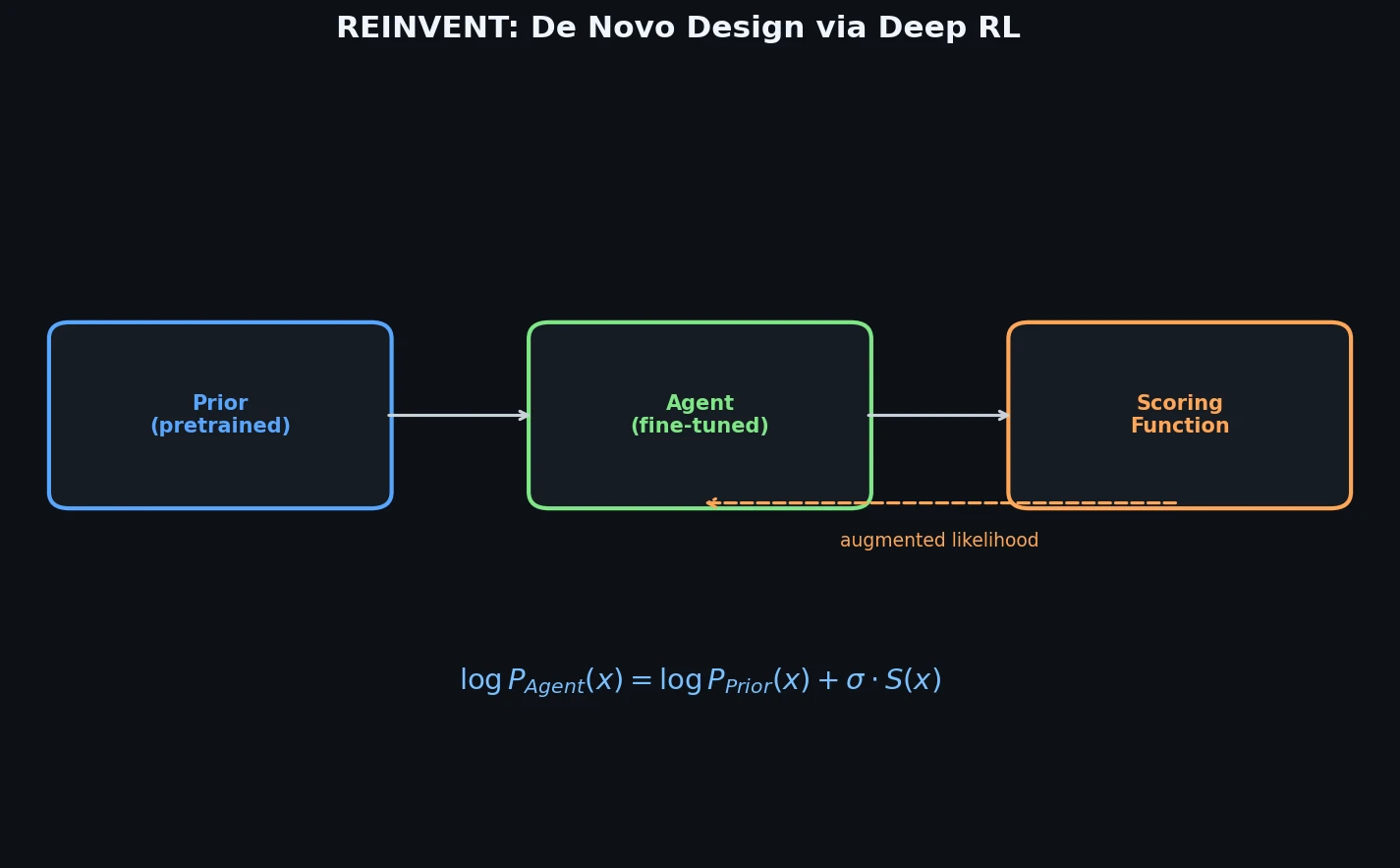
Introduces a policy-based reinforcement learning method that fine-tunes an RNN pre-trained on ChEMBL SMILES to generate molecules with specified desirable properties, using an augmented episodic likelihood that anchors the agent to its prior while optimizing a user-defined scoring function.
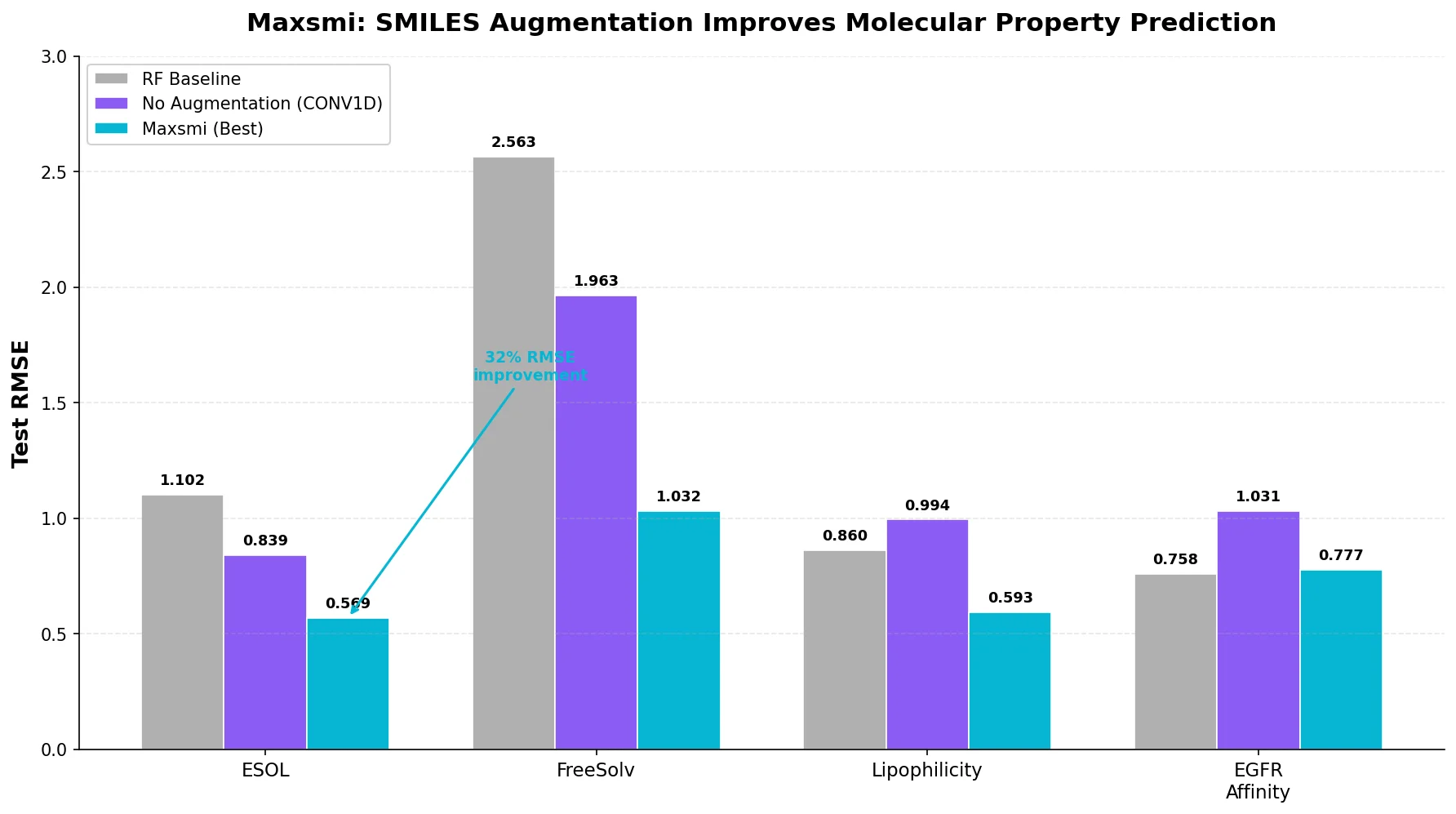
A systematic study of SMILES augmentation strategies for molecular property prediction, showing that augmentation consistently improves CNN and RNN performance and that prediction variance across SMILES correlates with model uncertainty.