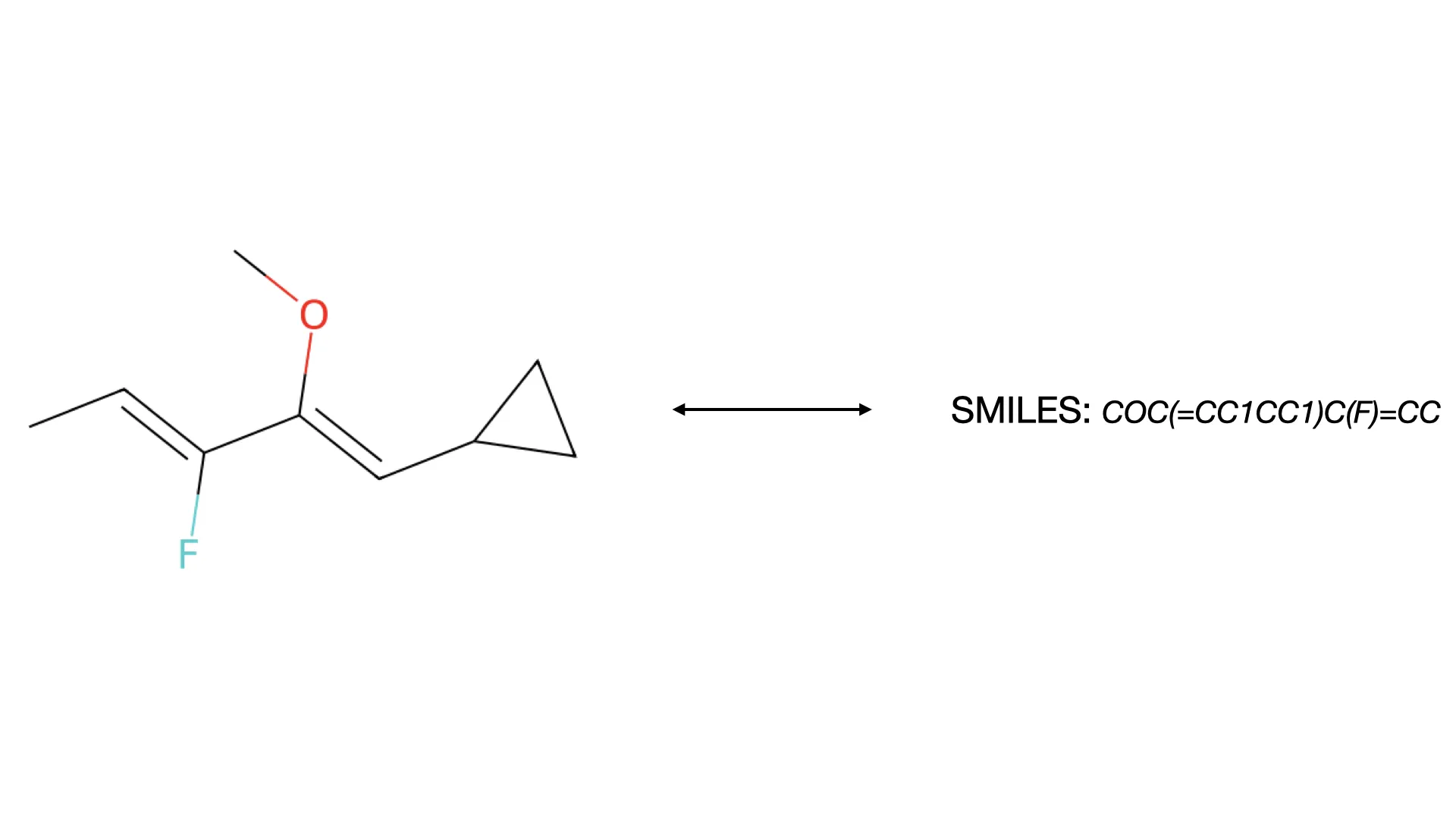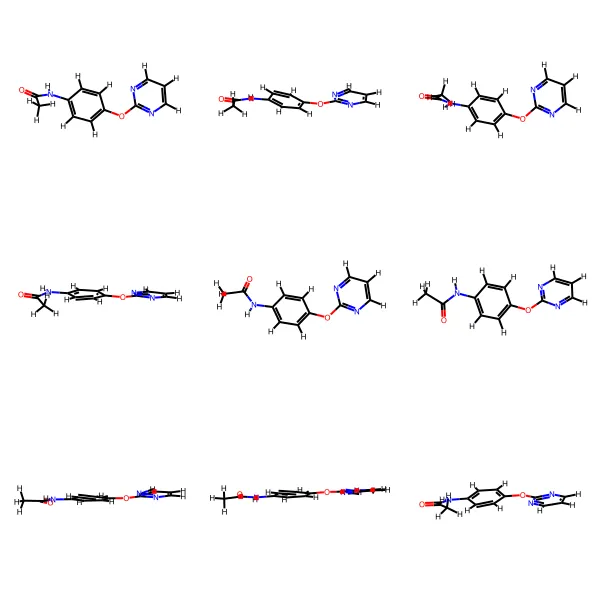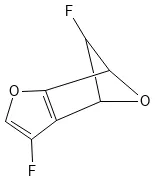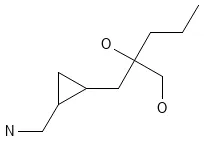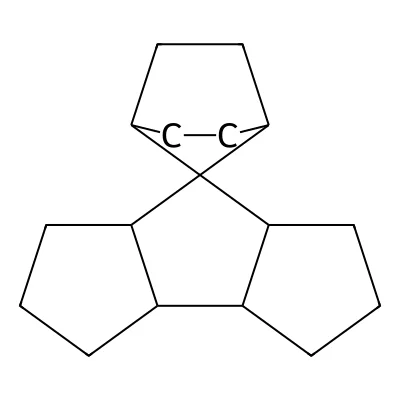
Deep Learning for Molecular Structure Extraction (2019)
This paper presents a two-stage deep learning pipeline to extract chemical structures from documents and convert them to SMILES strings. By training on large-scale synthetic data, the method overcomes the brittleness of rule-based systems and demonstrates high accuracy even on low-resolution and noisy input images.