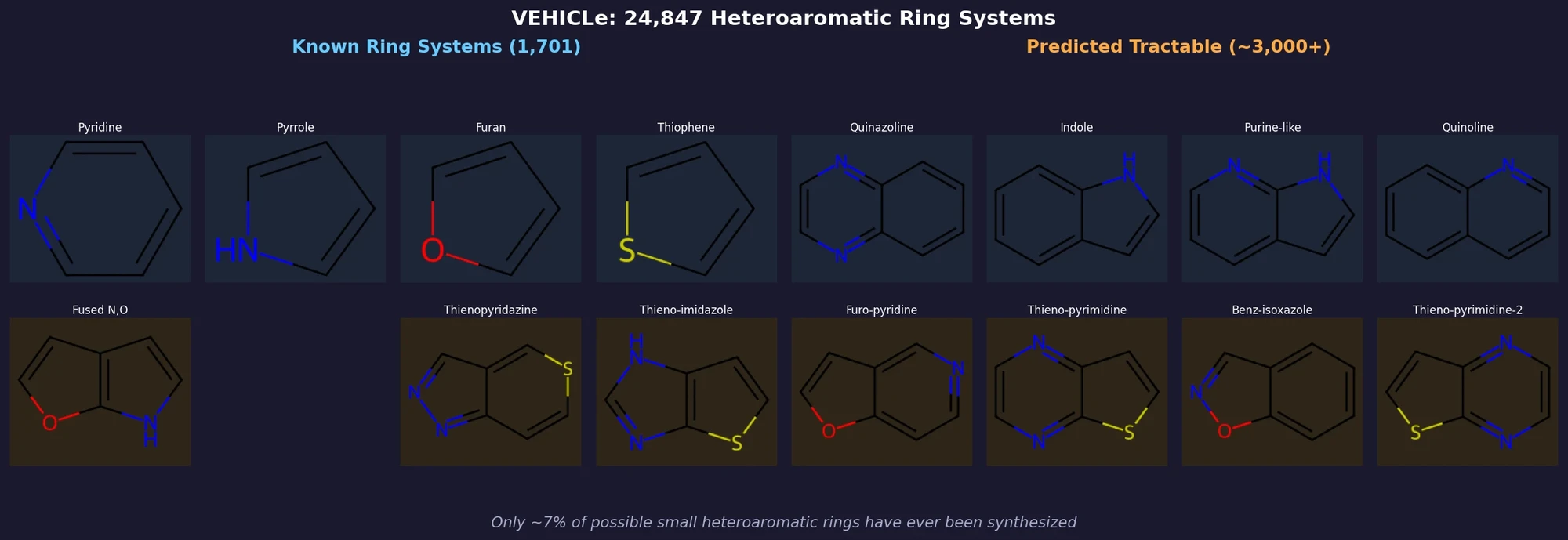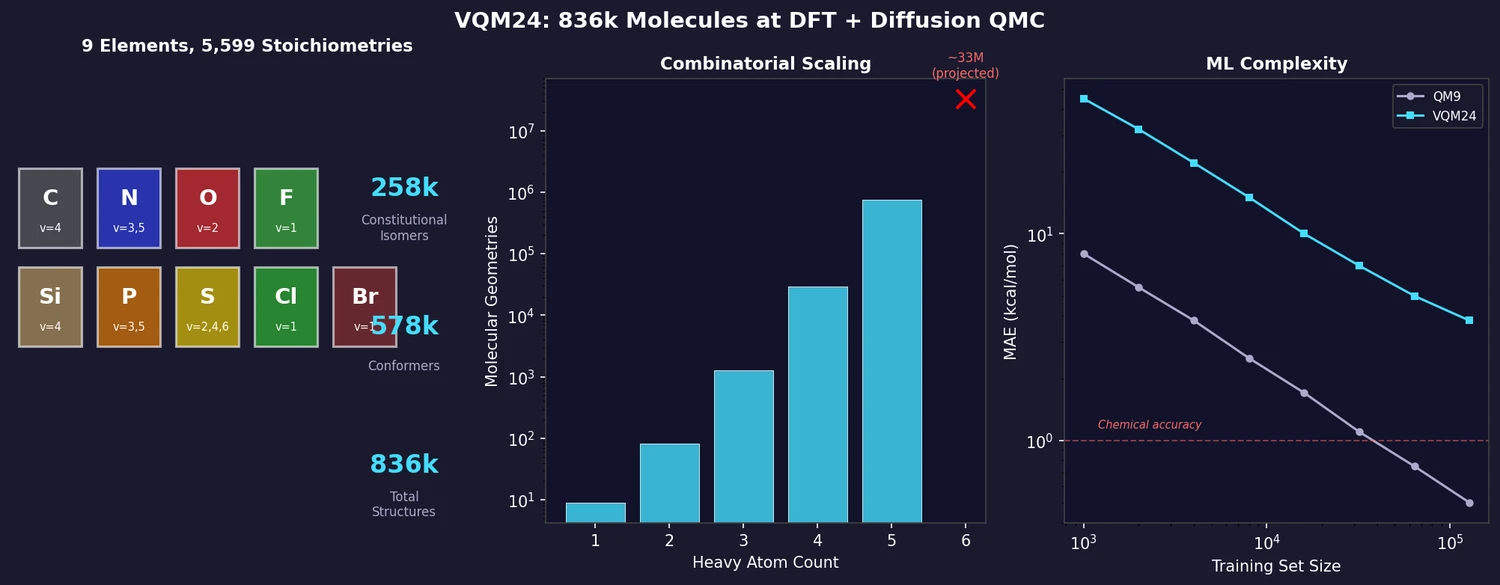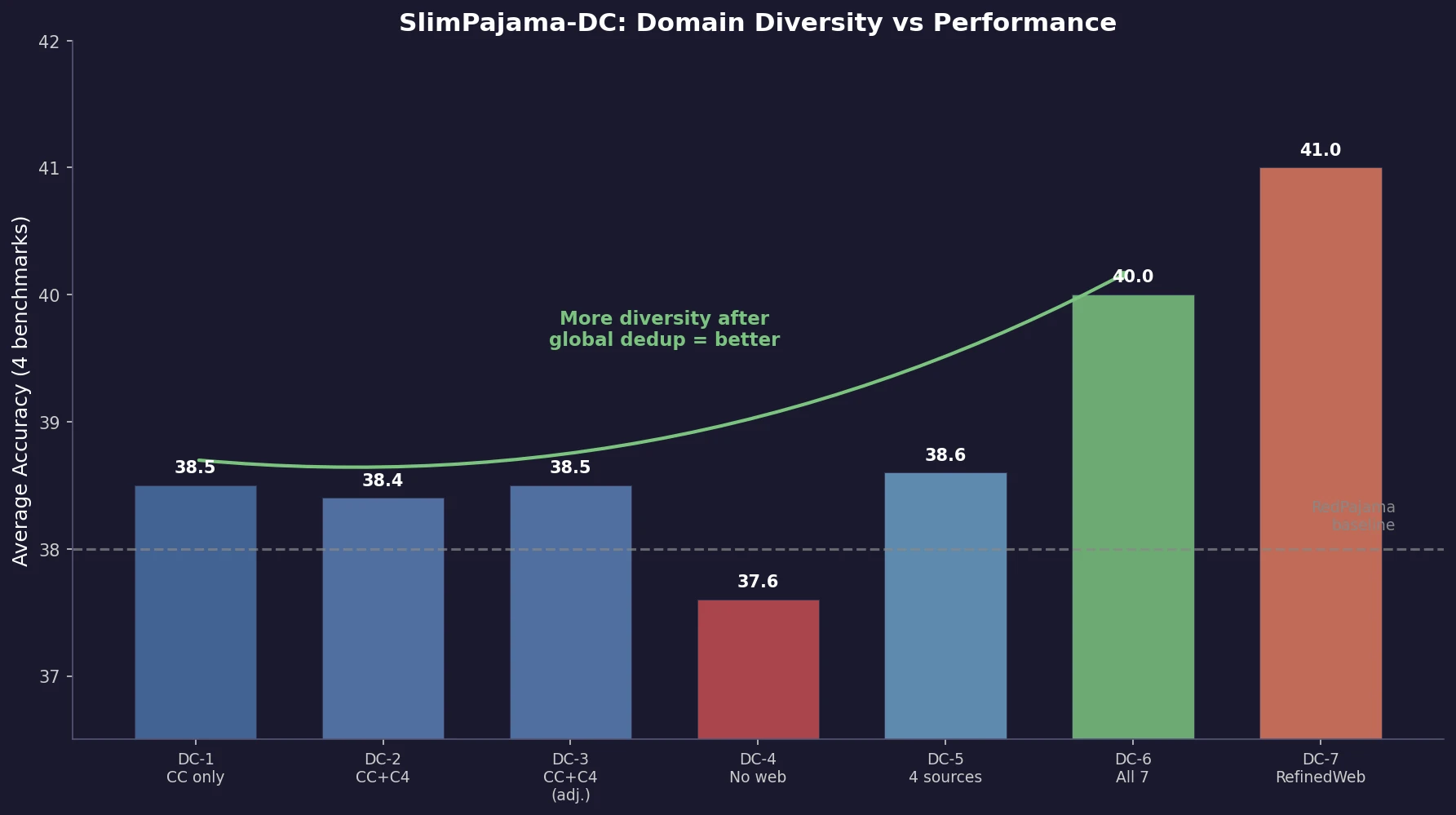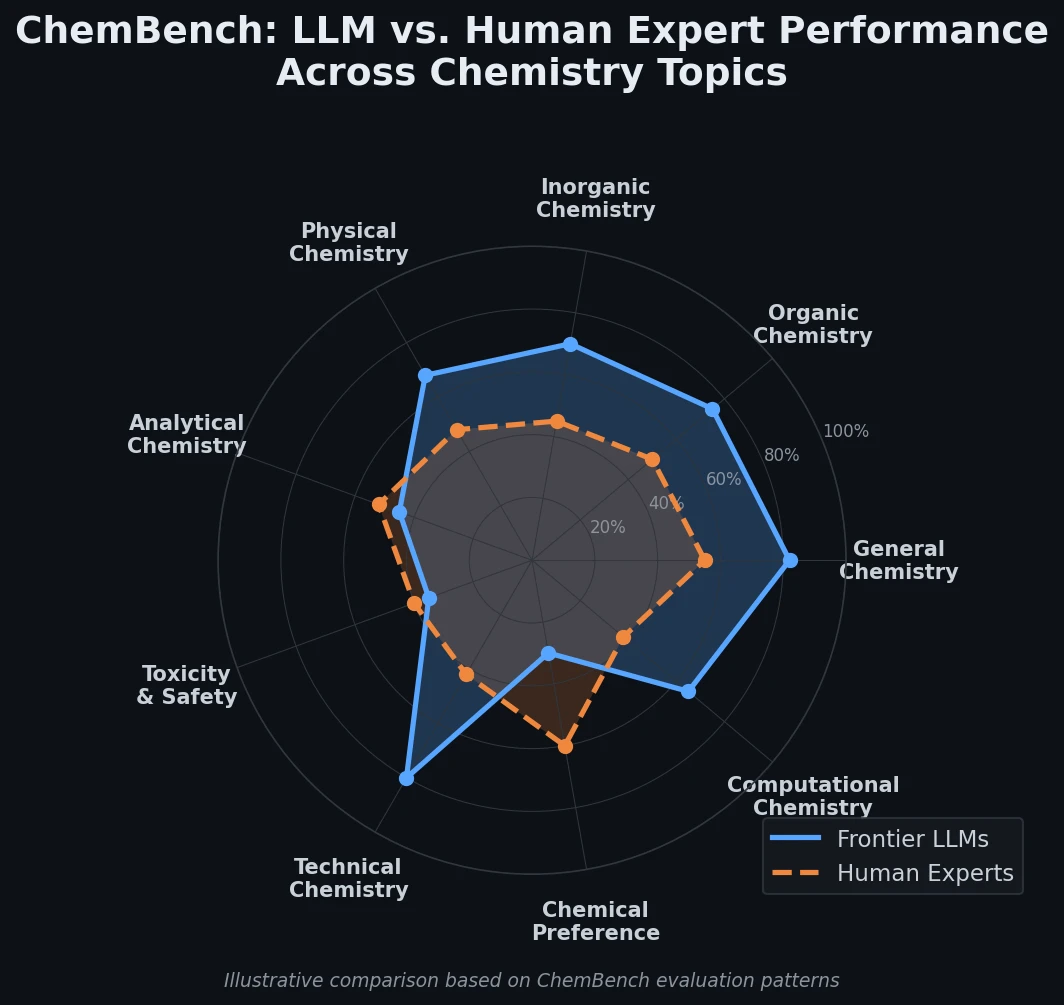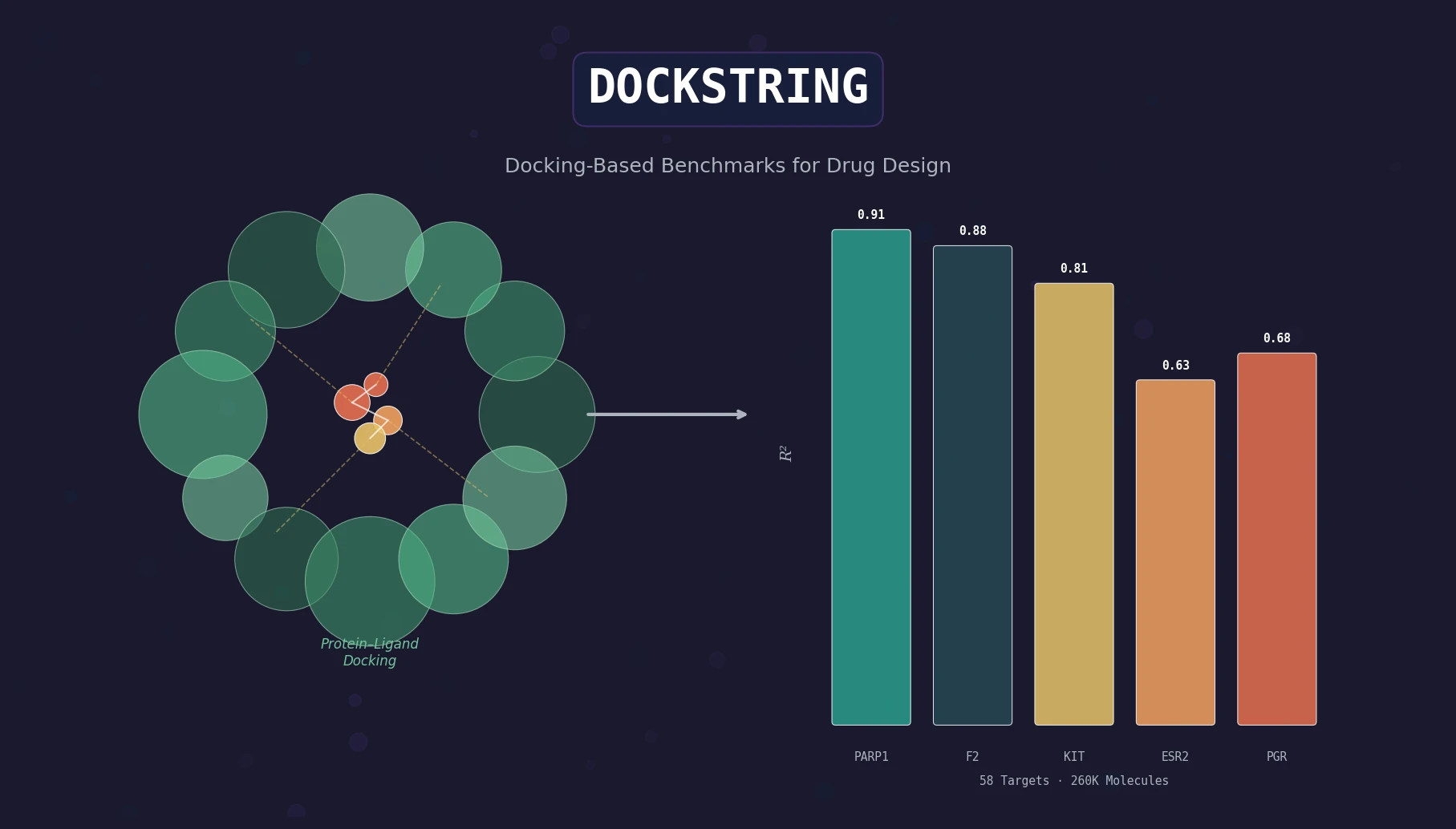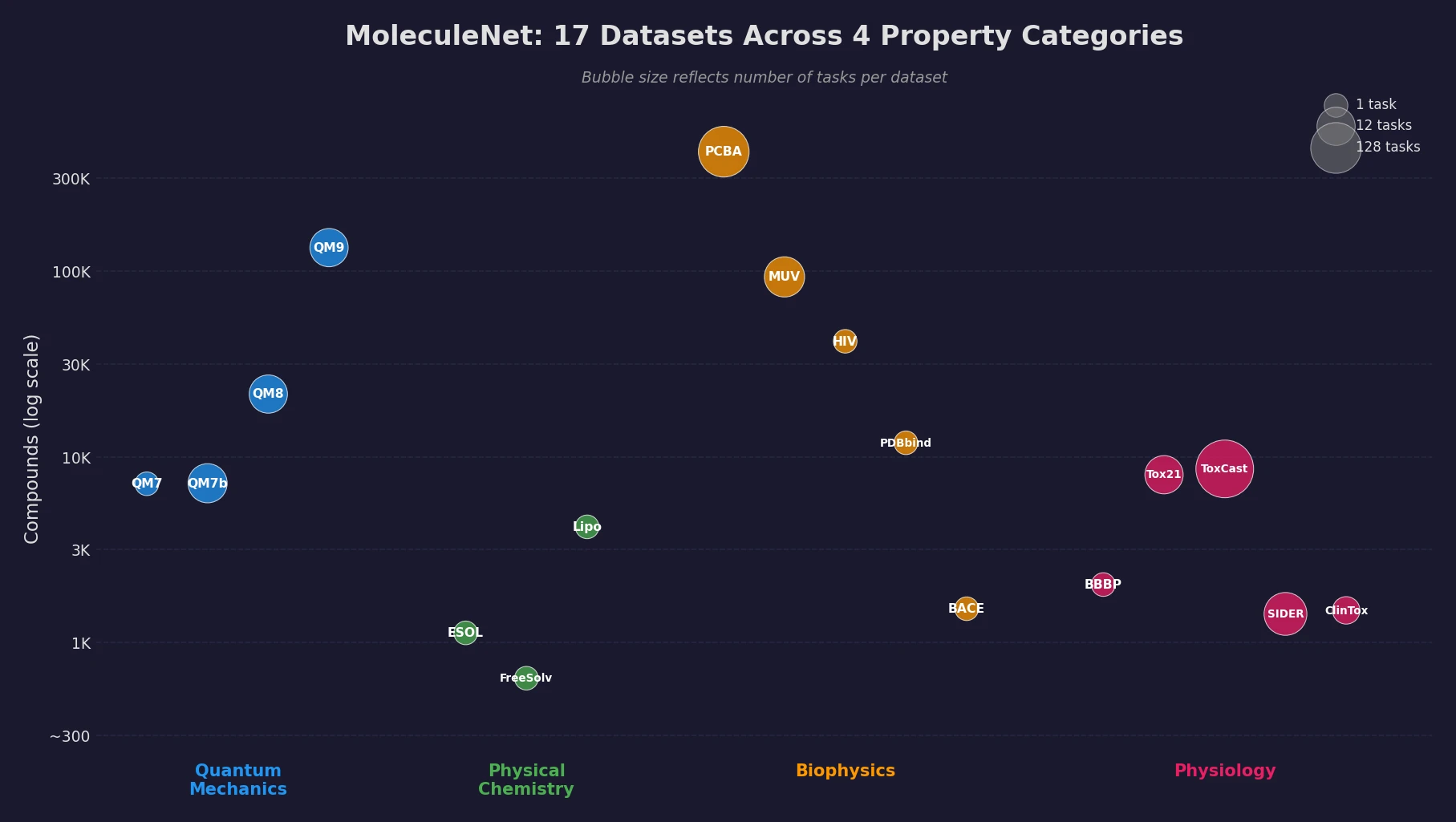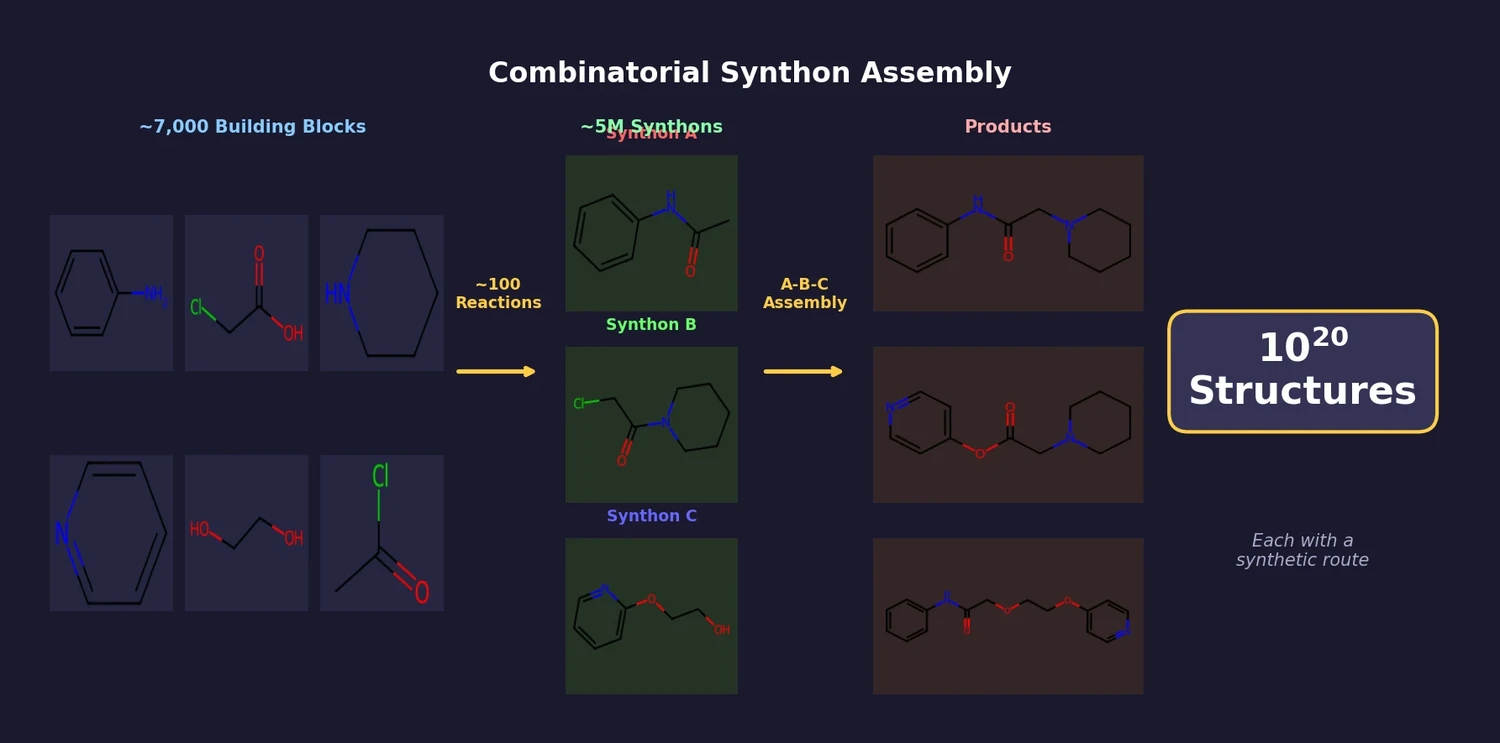
AllChem: Generating and Searching 10^20 Structures
AllChem generates ~5 million synthons by recursively applying ~100 reactions to ~7,000 building blocks, combinatorially representing up to 10^20 complete structures with an A-B-C topology. Topomer shape similarity enables efficient searching of this space, and every hit comes with a proposed synthetic route.