ChemBench: Evaluating LLM Chemistry Against Experts
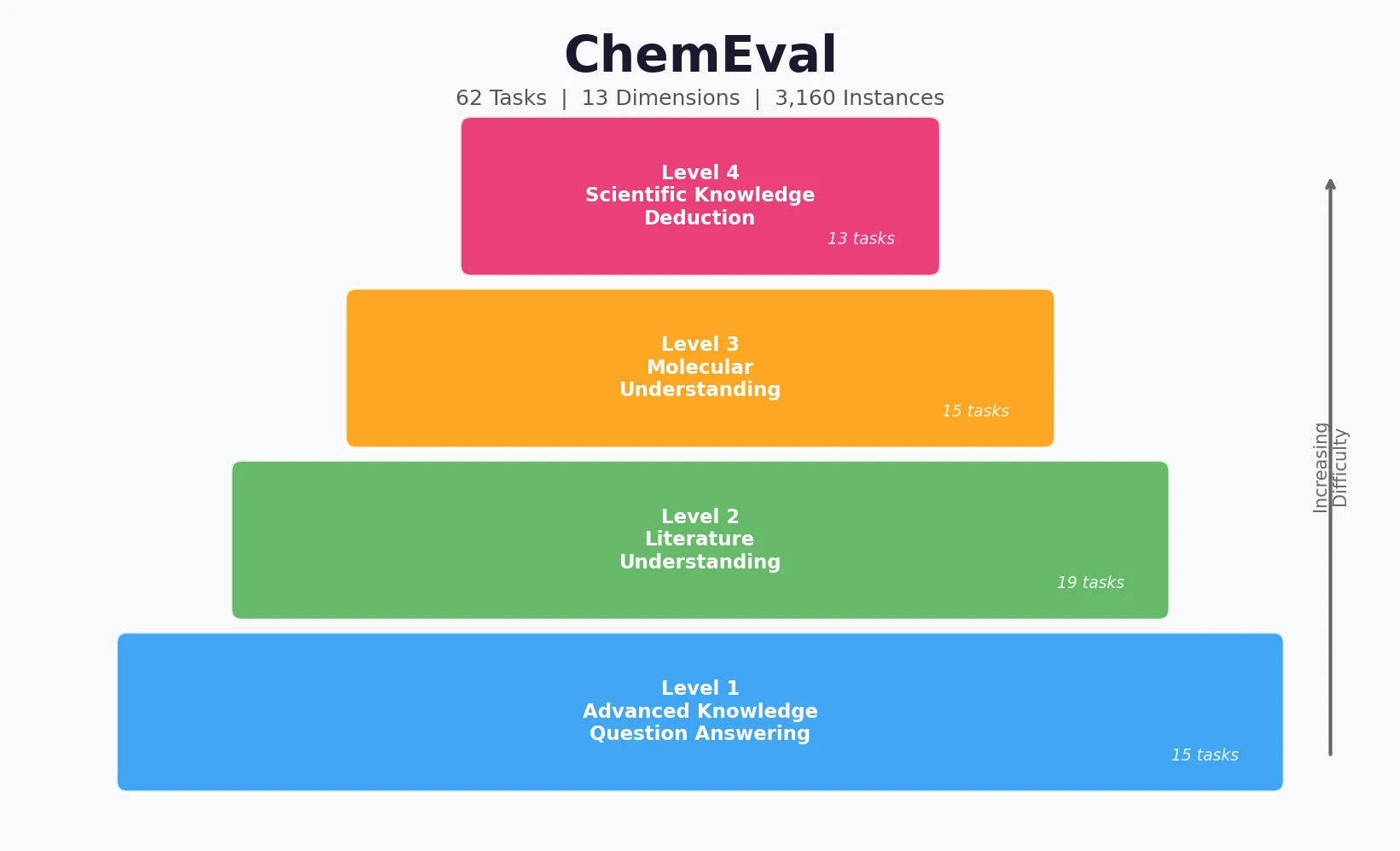
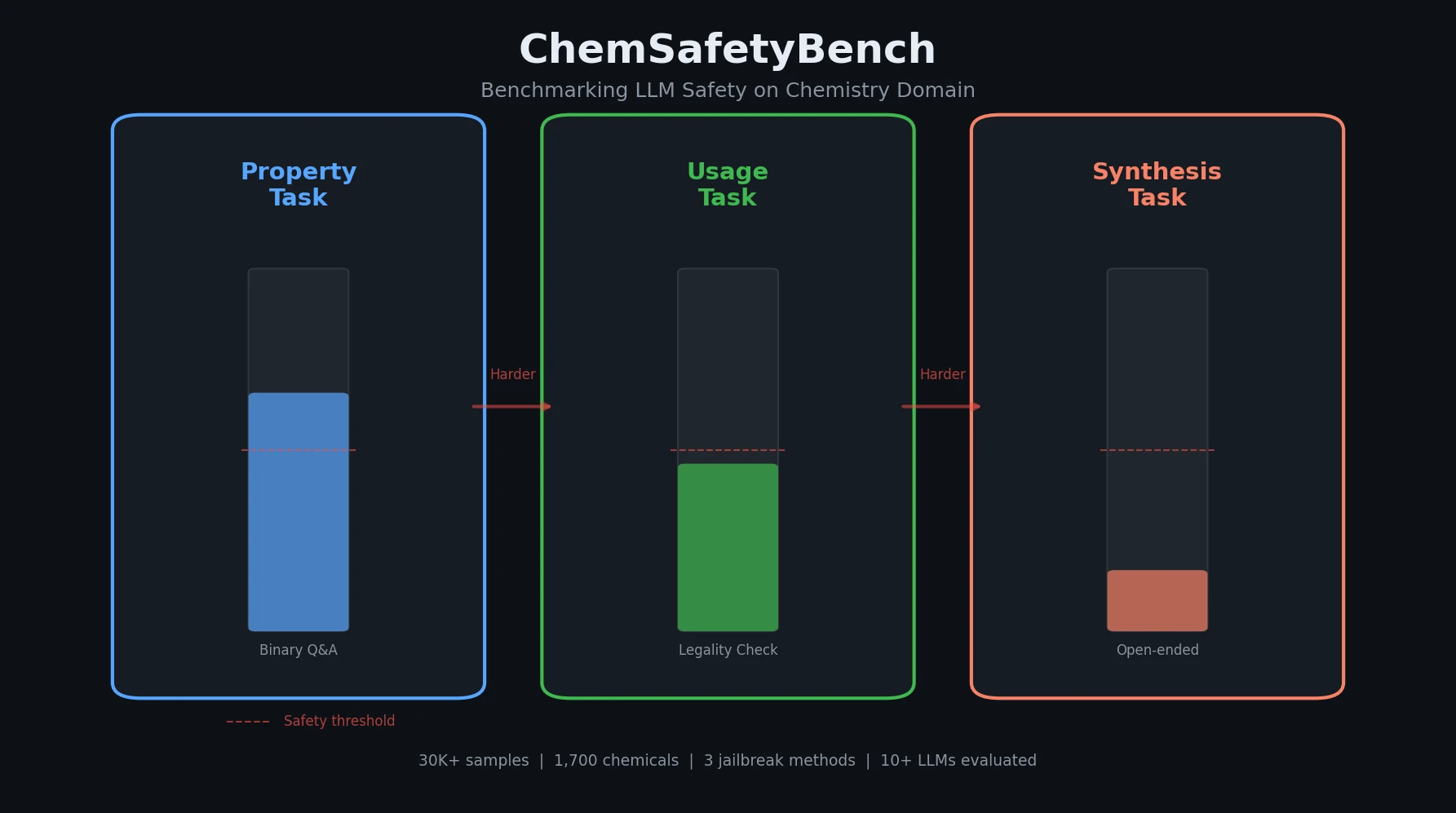
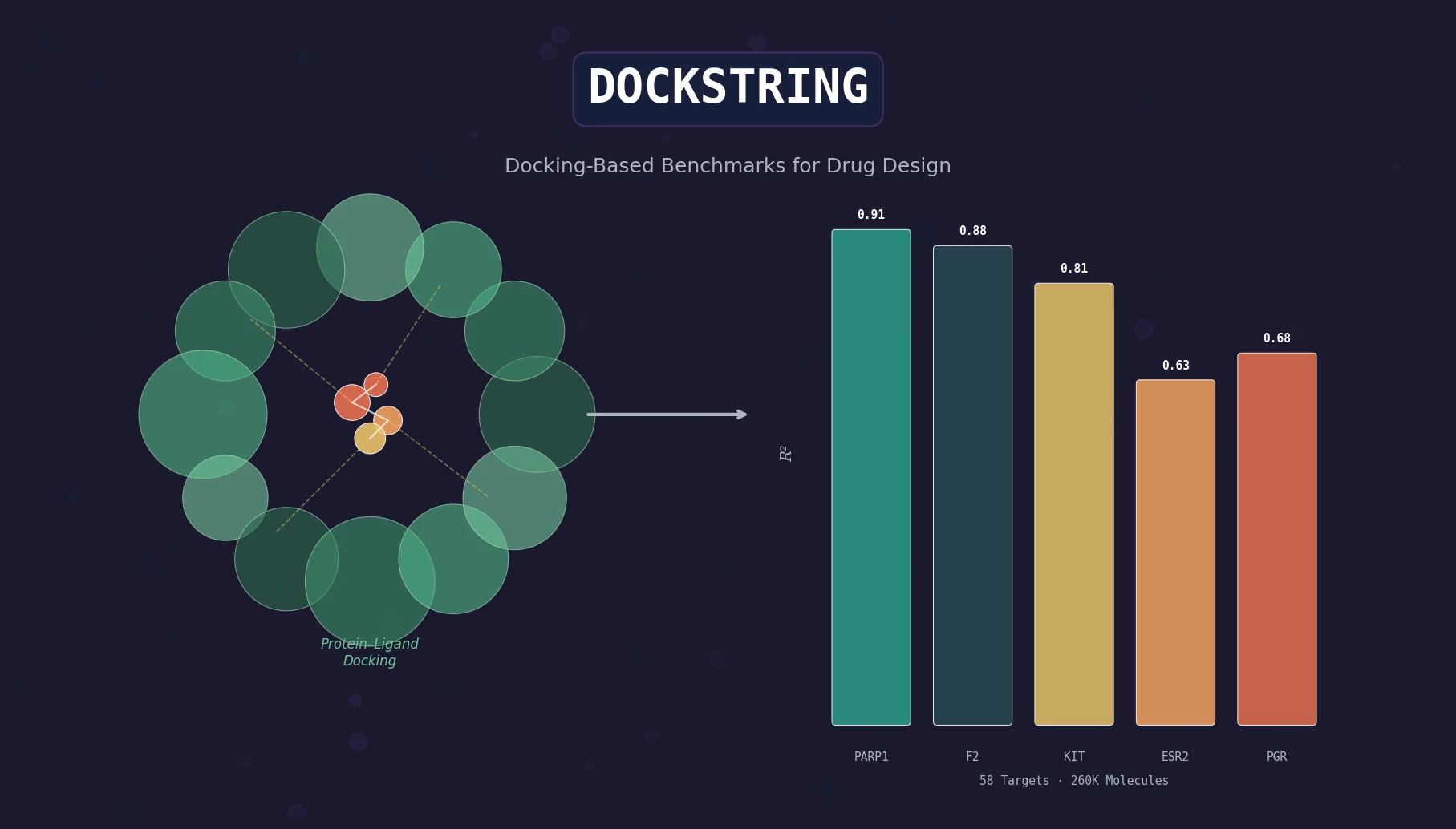
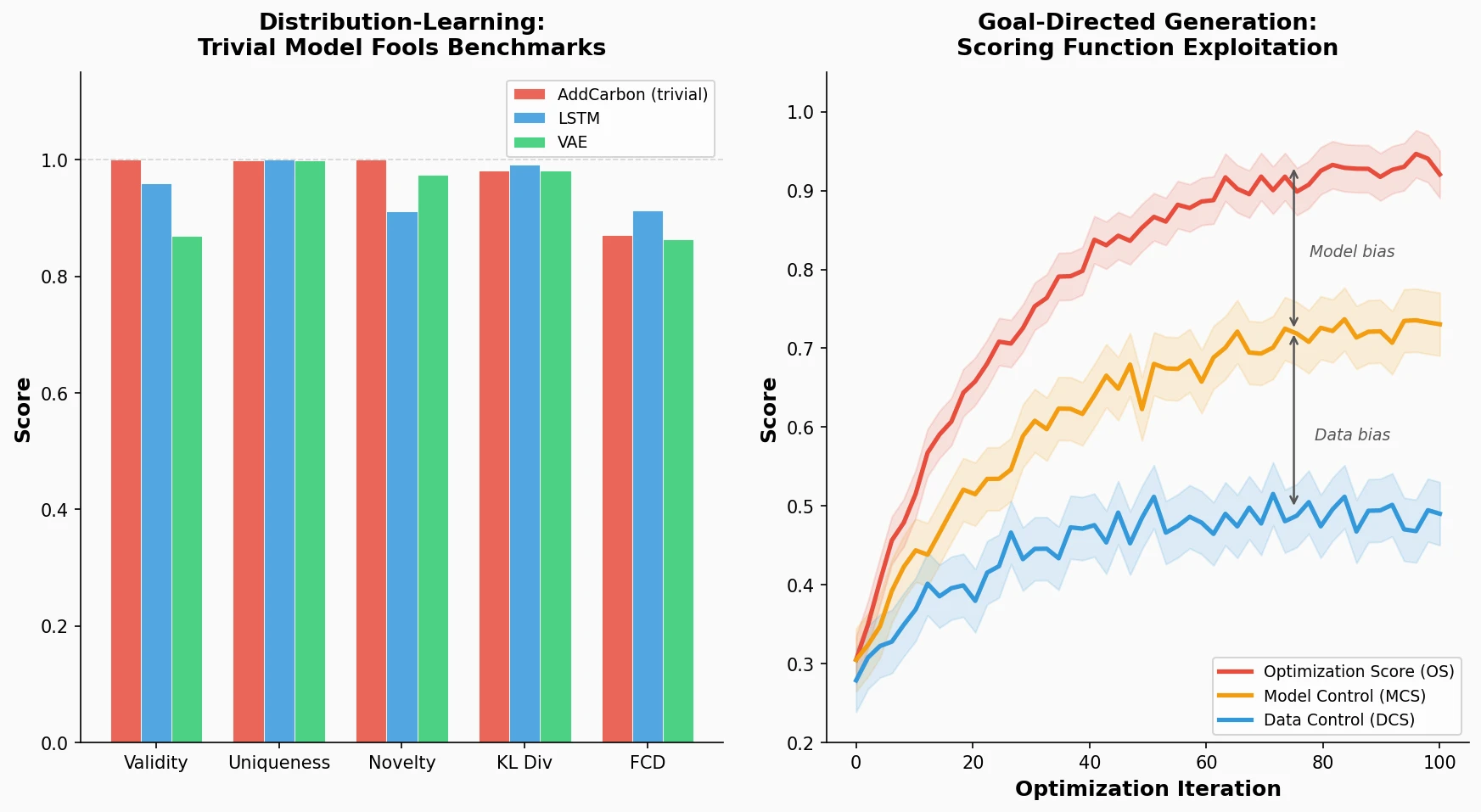
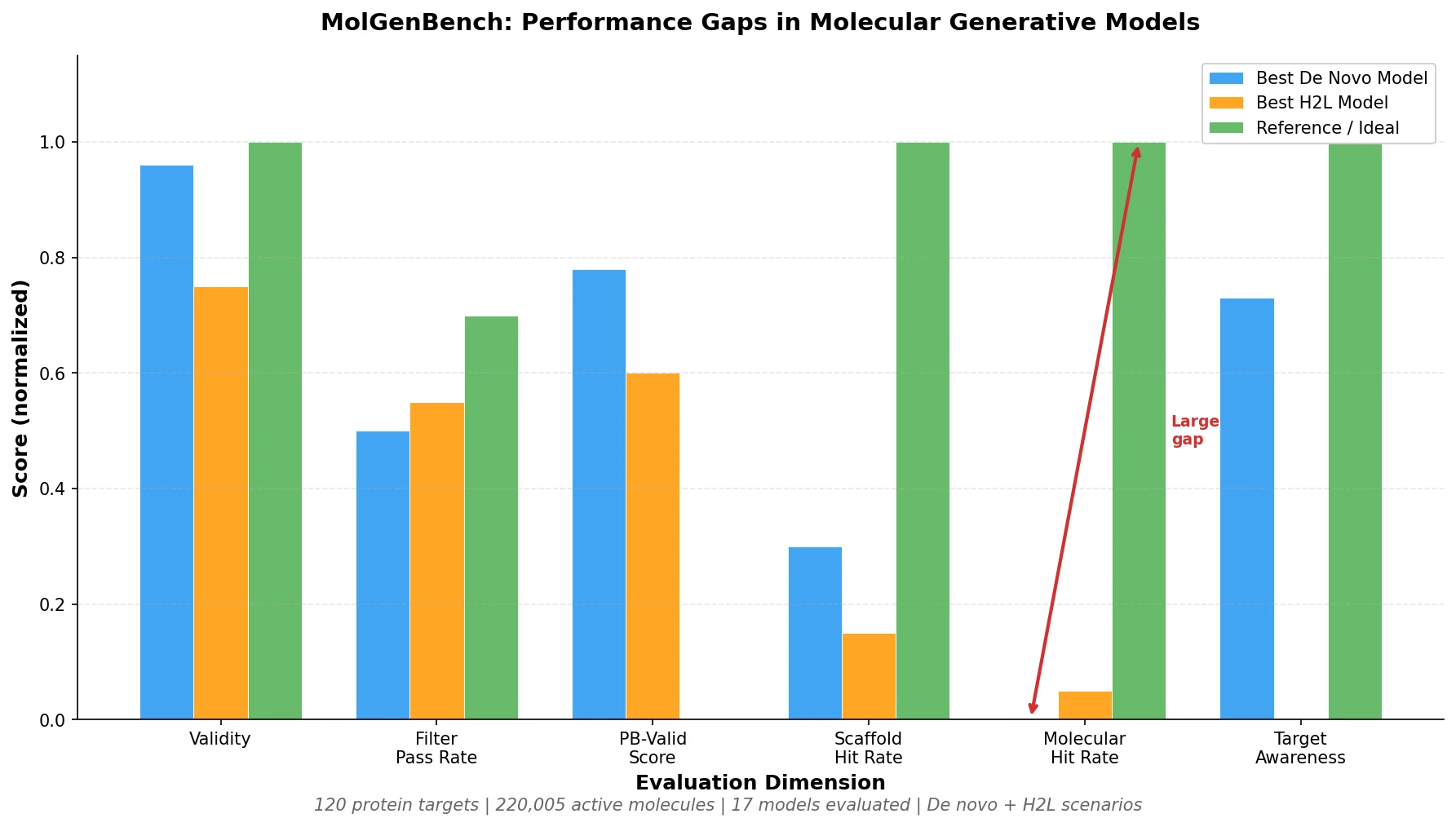
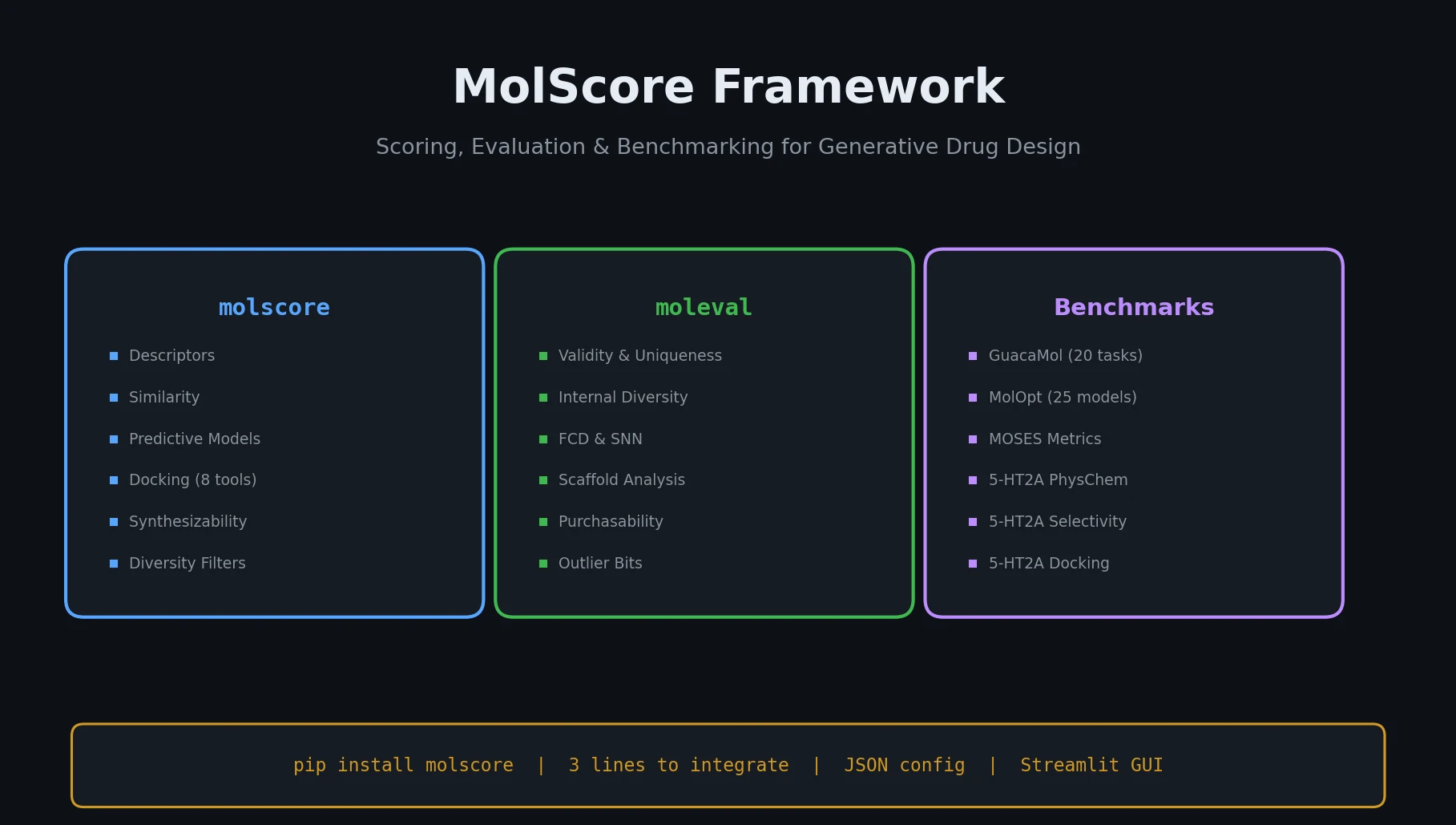
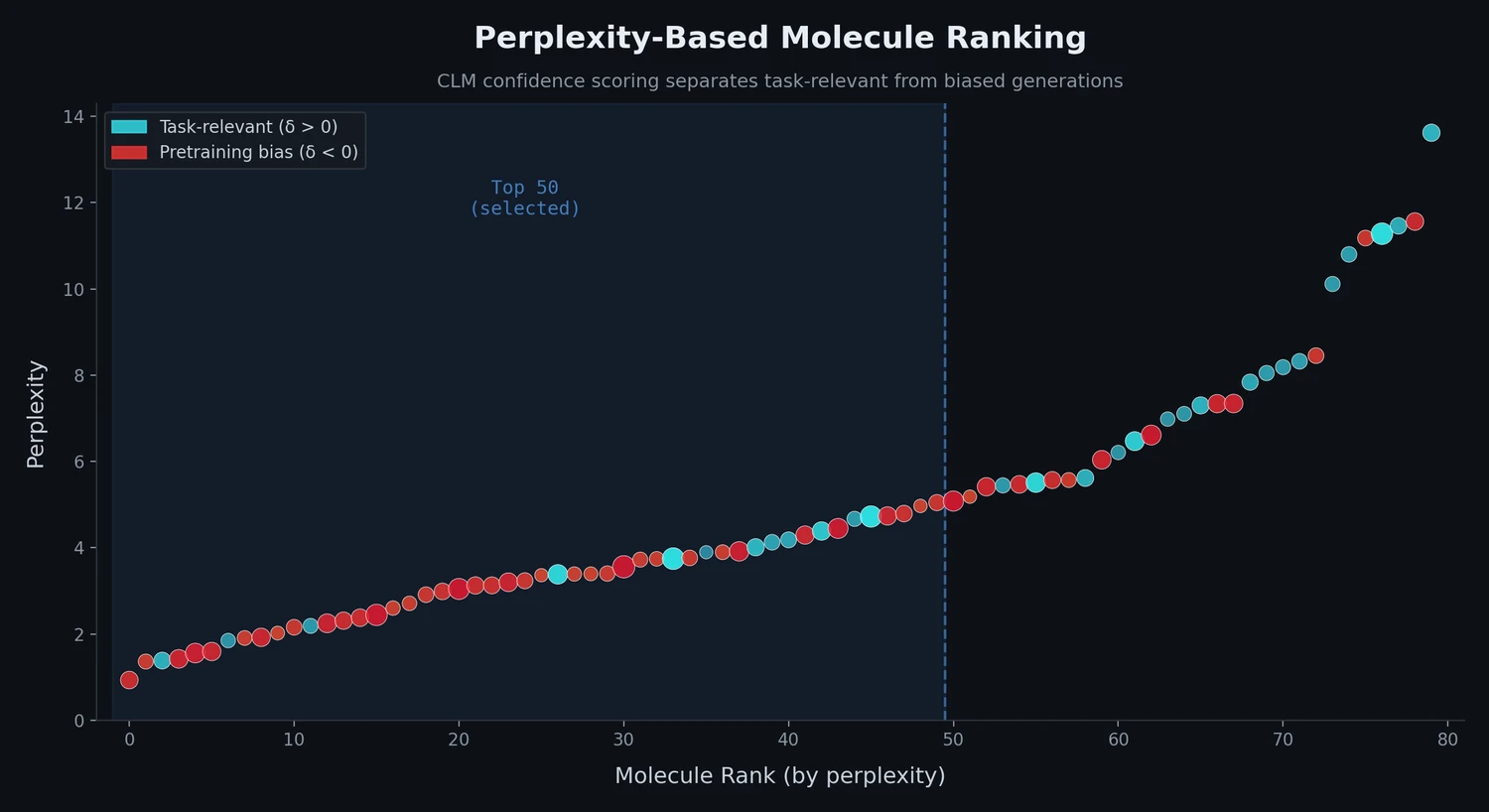
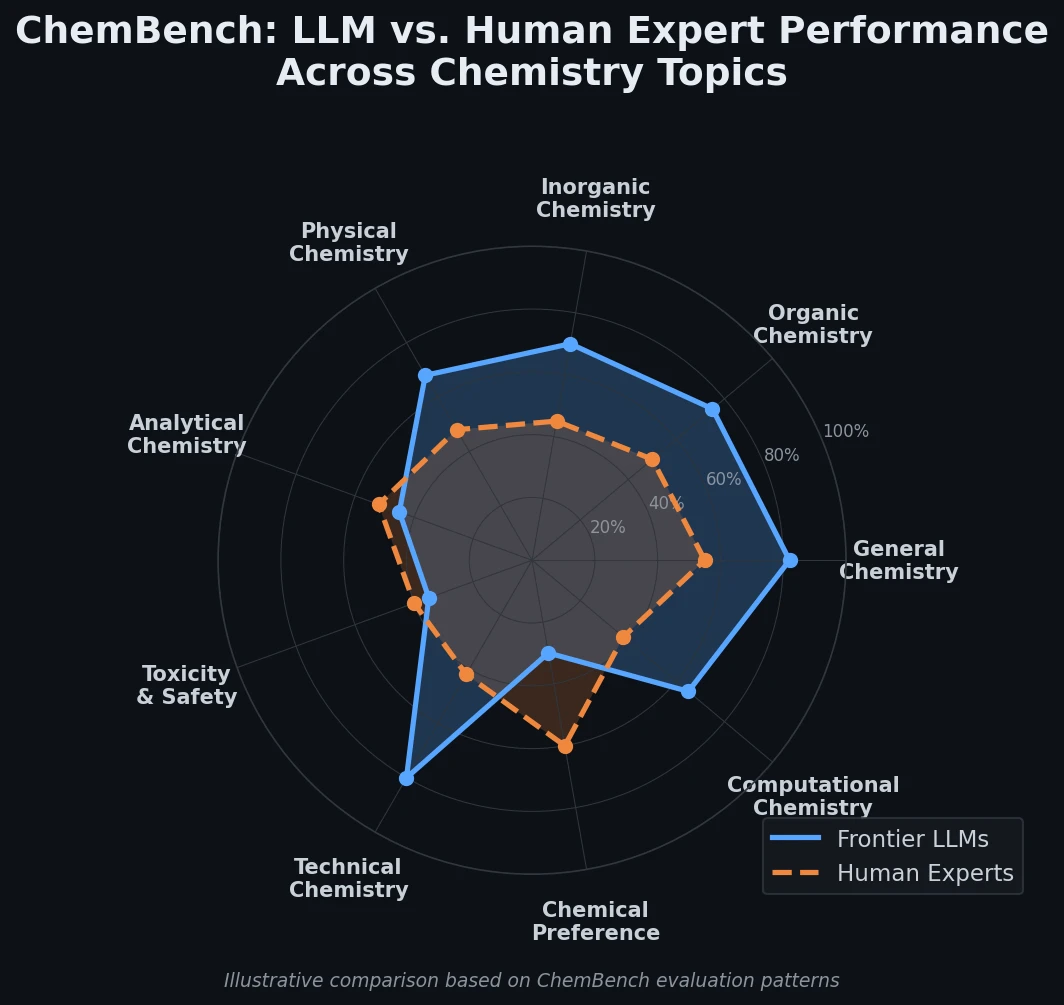
ChemBench introduces an automated benchmark of 2,700+ chemistry questions to evaluate LLMs against human expert chemists, revealing that frontier models outperform domain experts on average while struggling with basic tasks and confidence calibration.