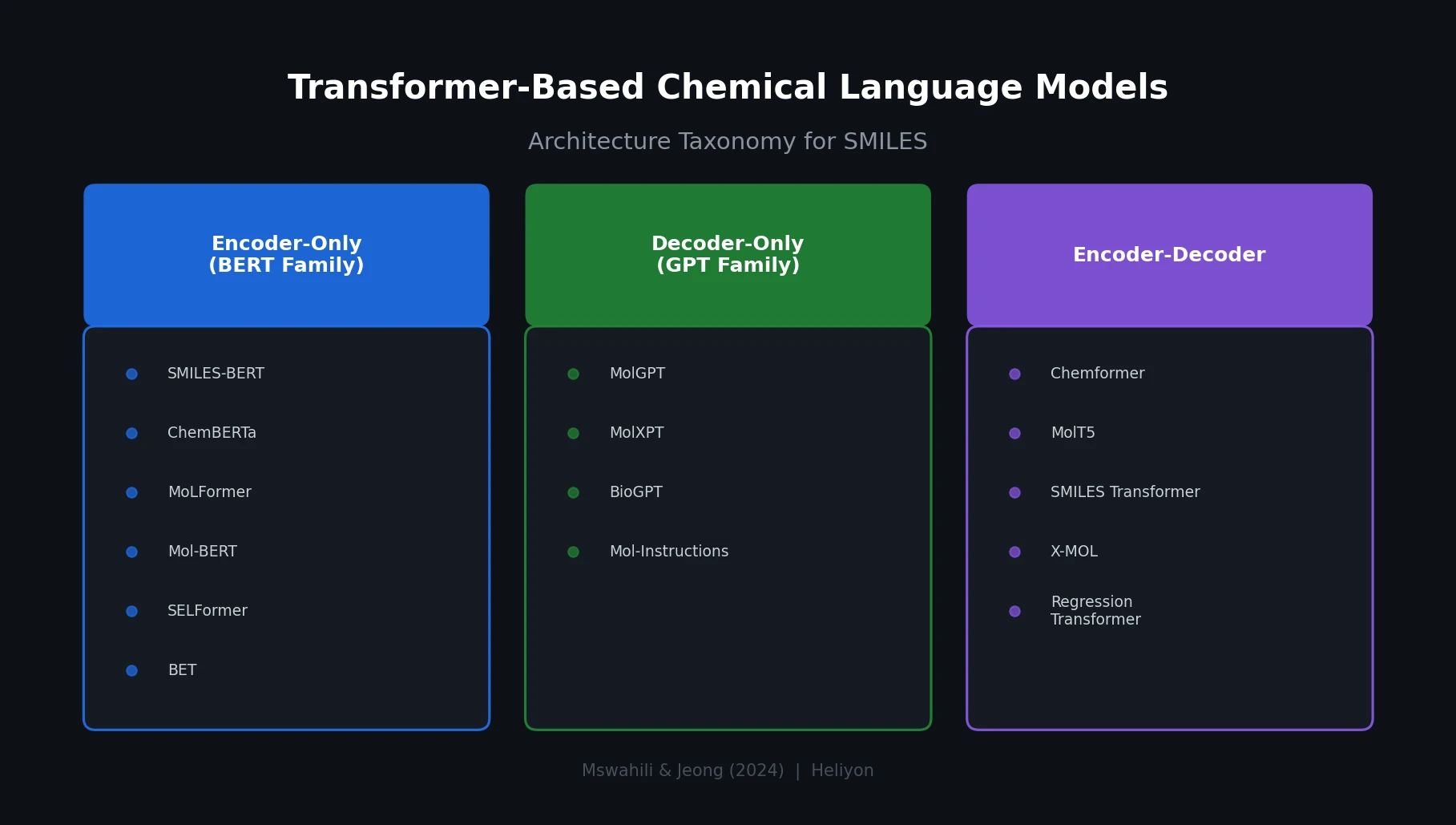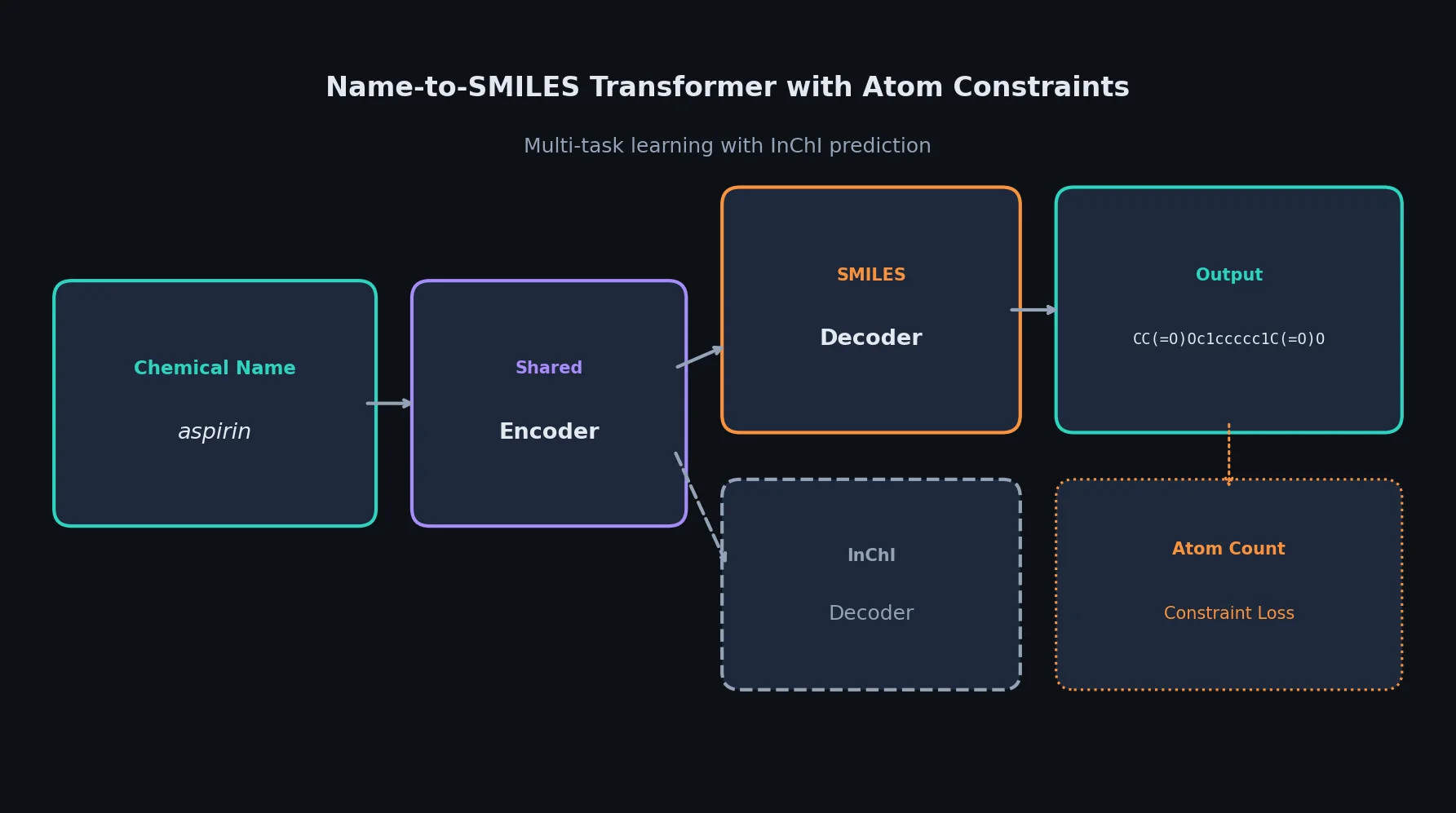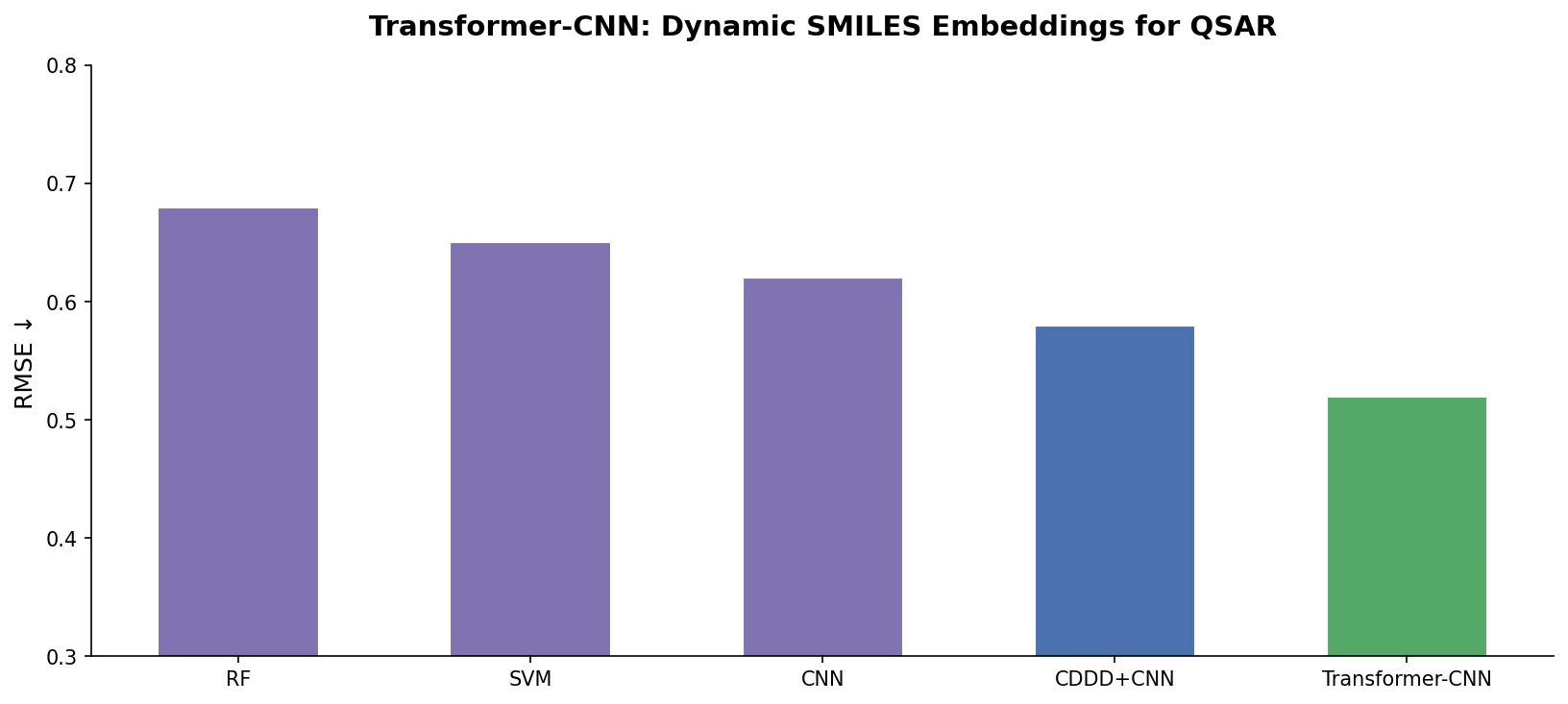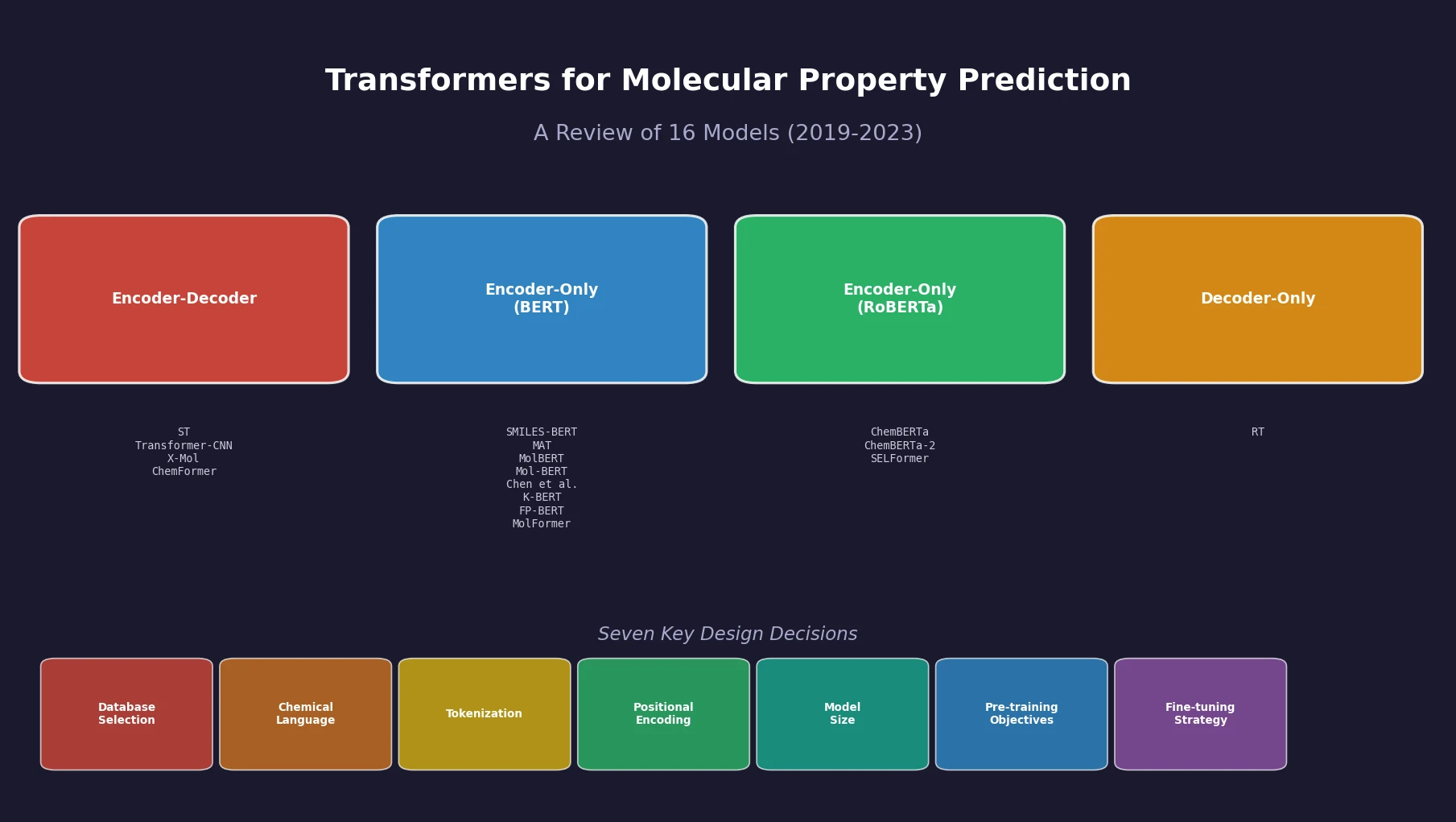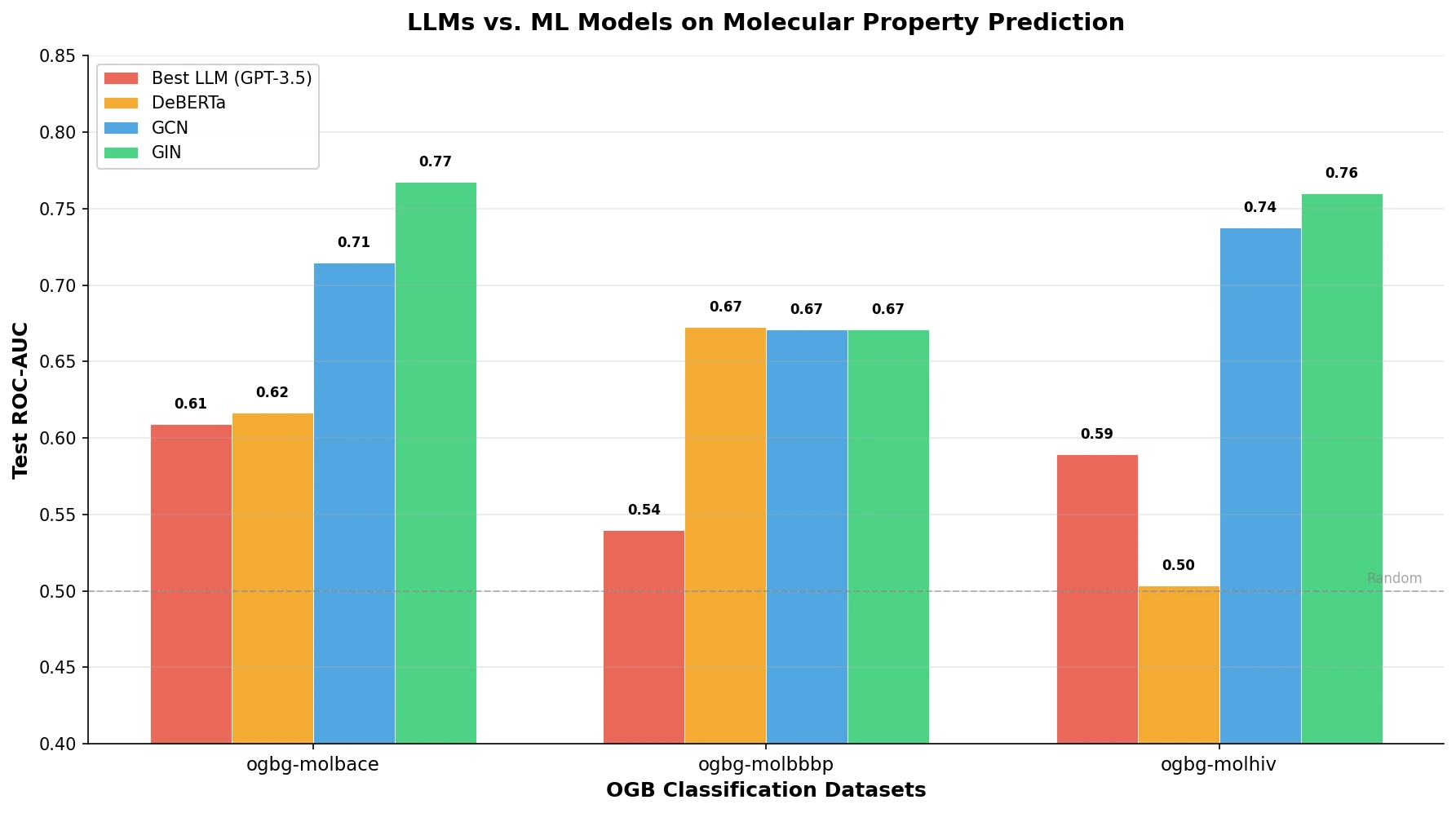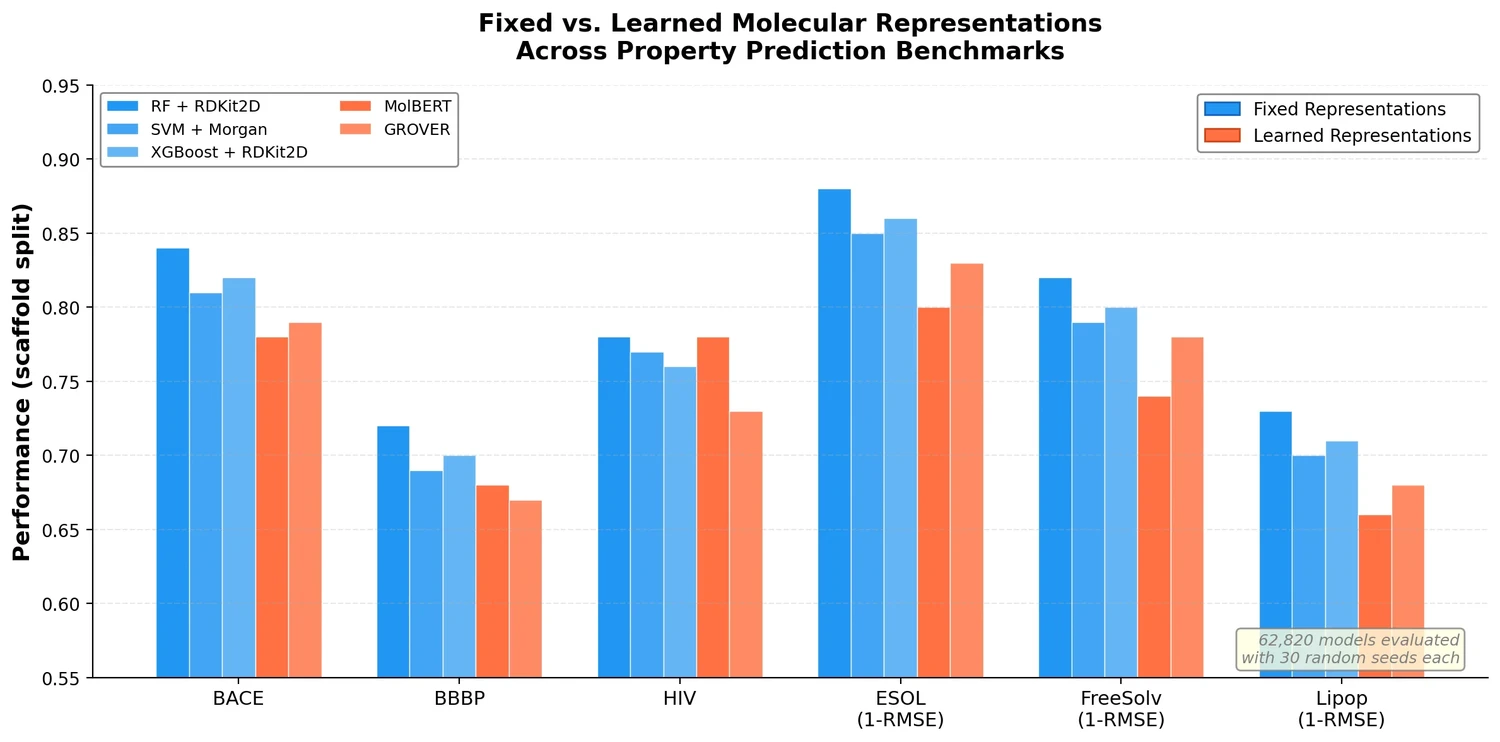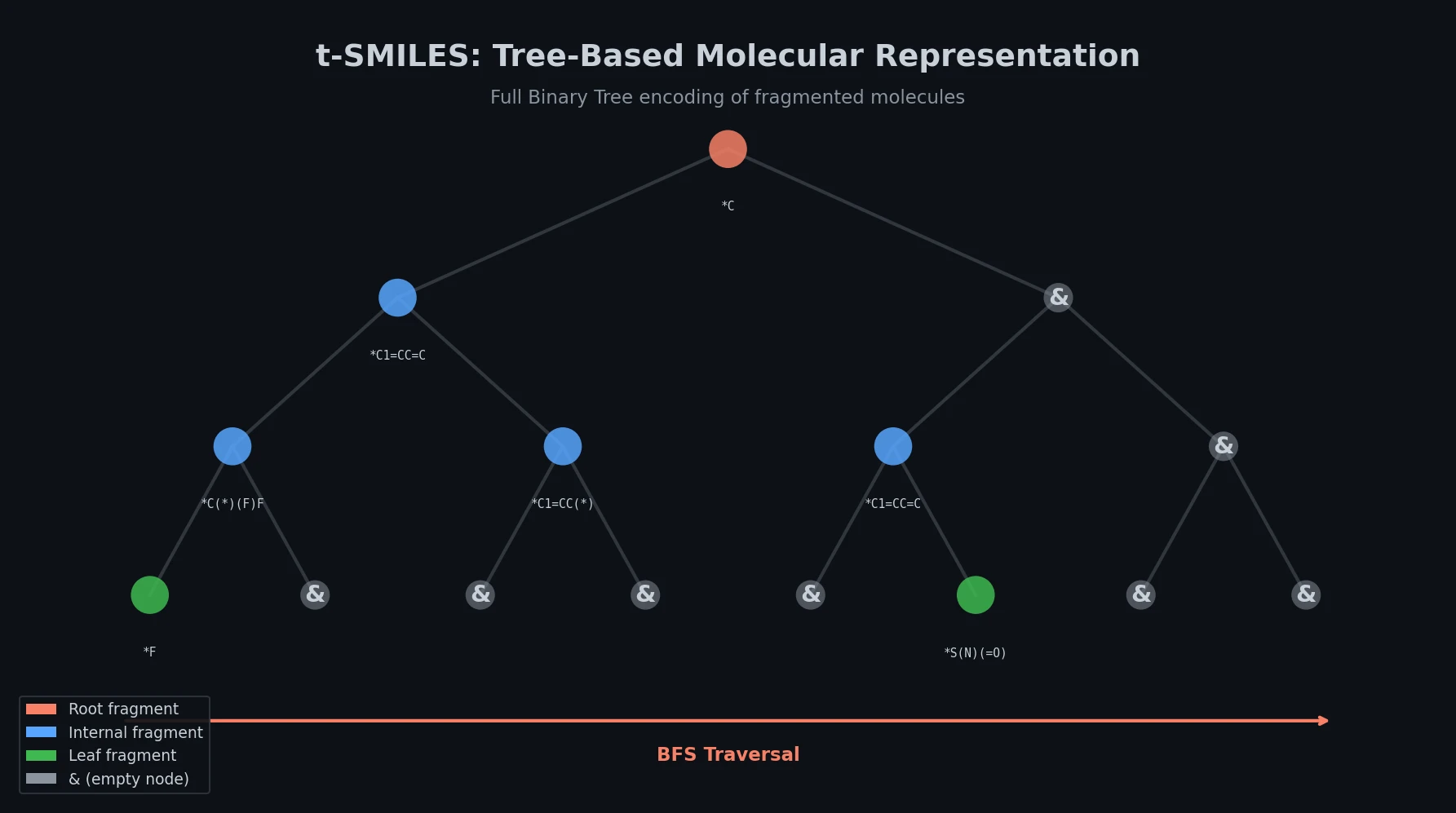
t-SMILES: Tree-Based Fragment Molecular Encoding
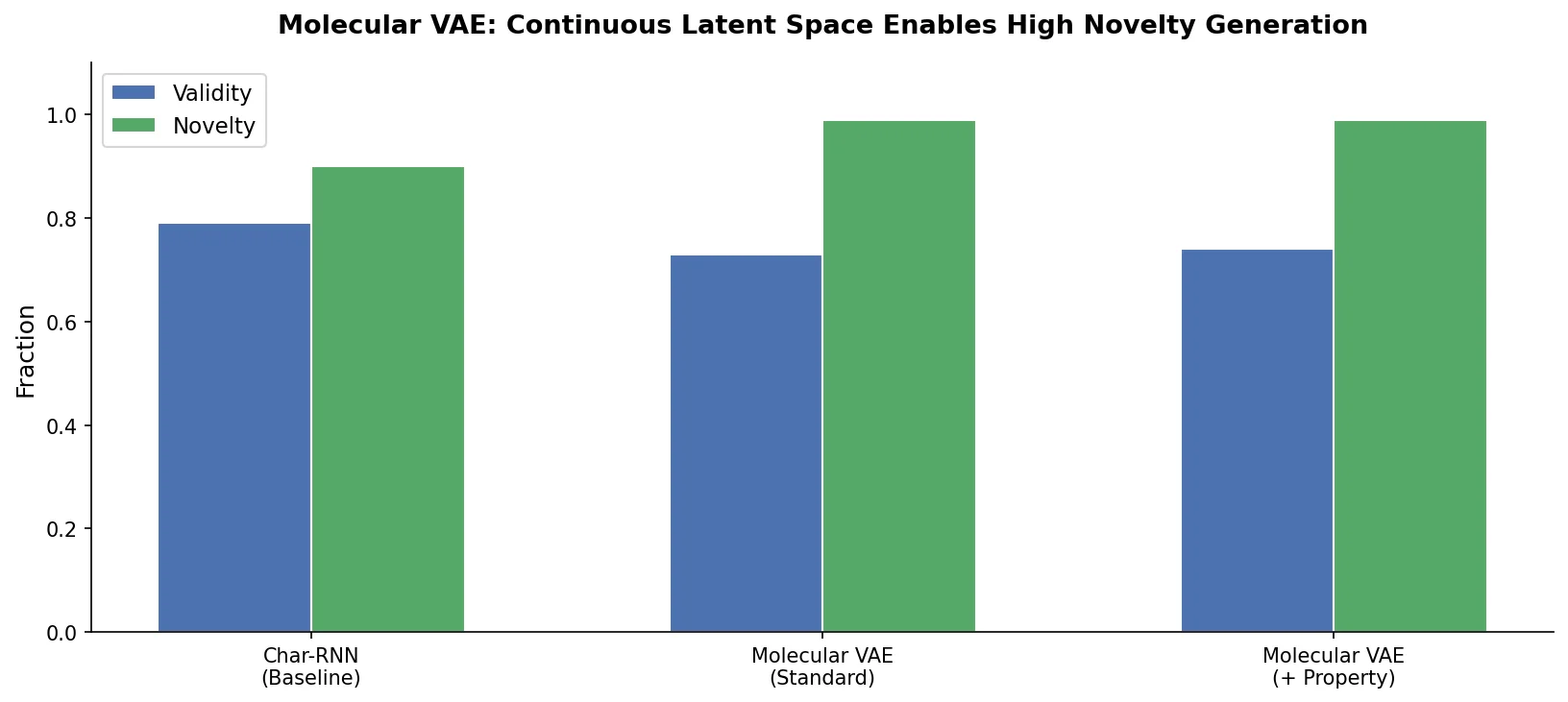
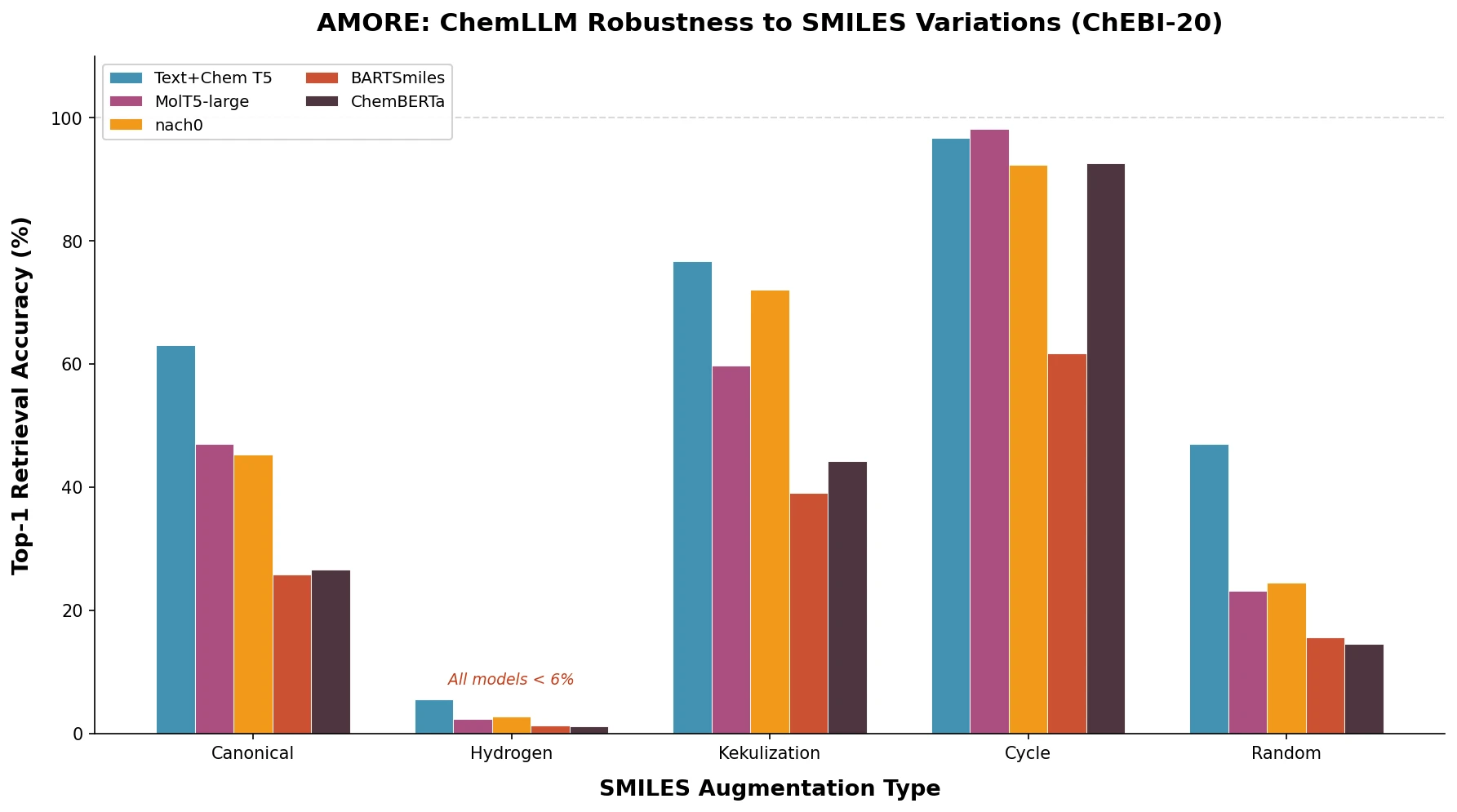
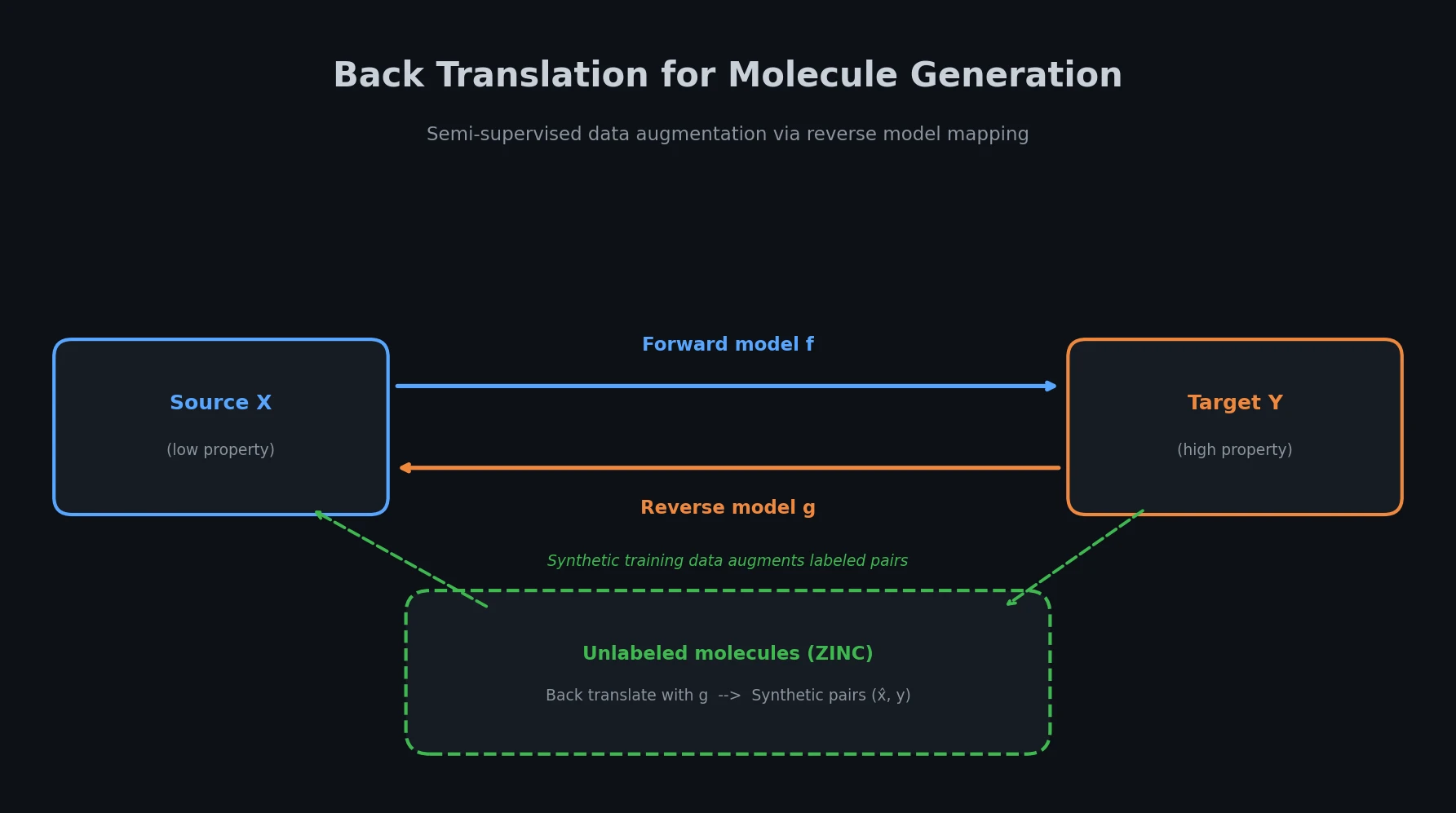
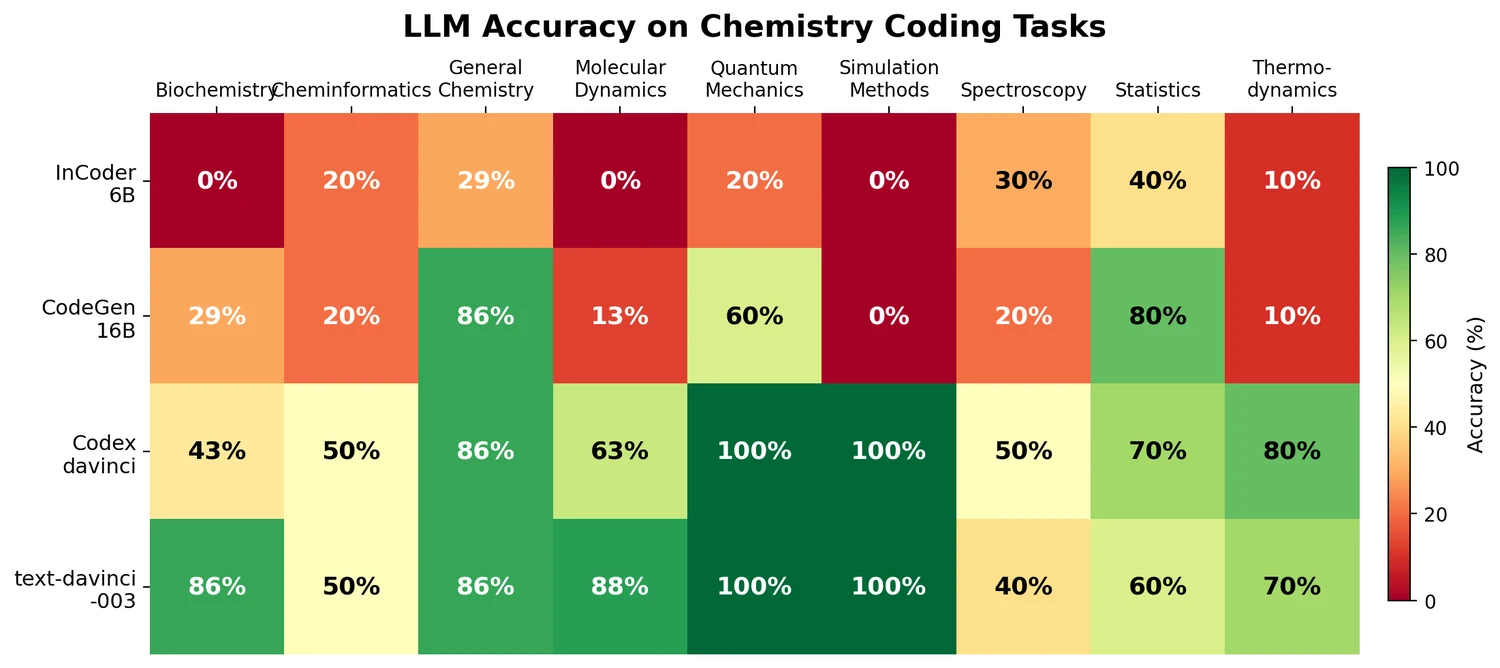
t-SMILES represents molecules by fragmenting them into substructures, building full binary trees, and traversing them breadth-first to produce SMILES-type strings that reduce nesting depth and outperform SMILES, DeepSMILES, and SELFIES on generation benchmarks.