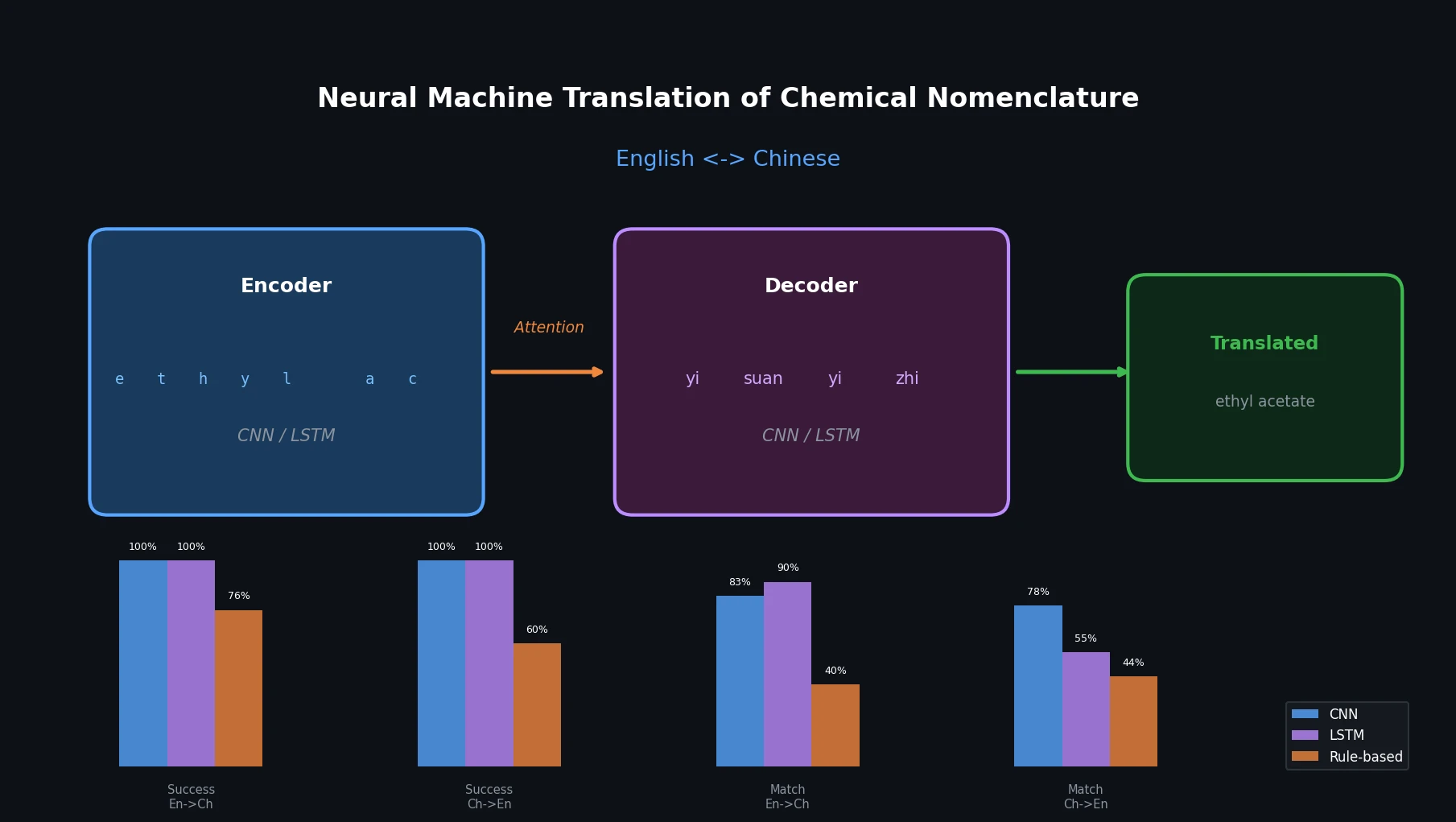
Neural Machine Translation of Chemical Nomenclature
This paper applies character-level CNN and LSTM encoder-decoder networks to translate chemical names between English and Chinese, comparing them against an existing rule-based tool.
This paper applies character-level CNN and LSTM encoder-decoder networks to translate chemical names between English and Chinese, comparing them against an existing rule-based tool.
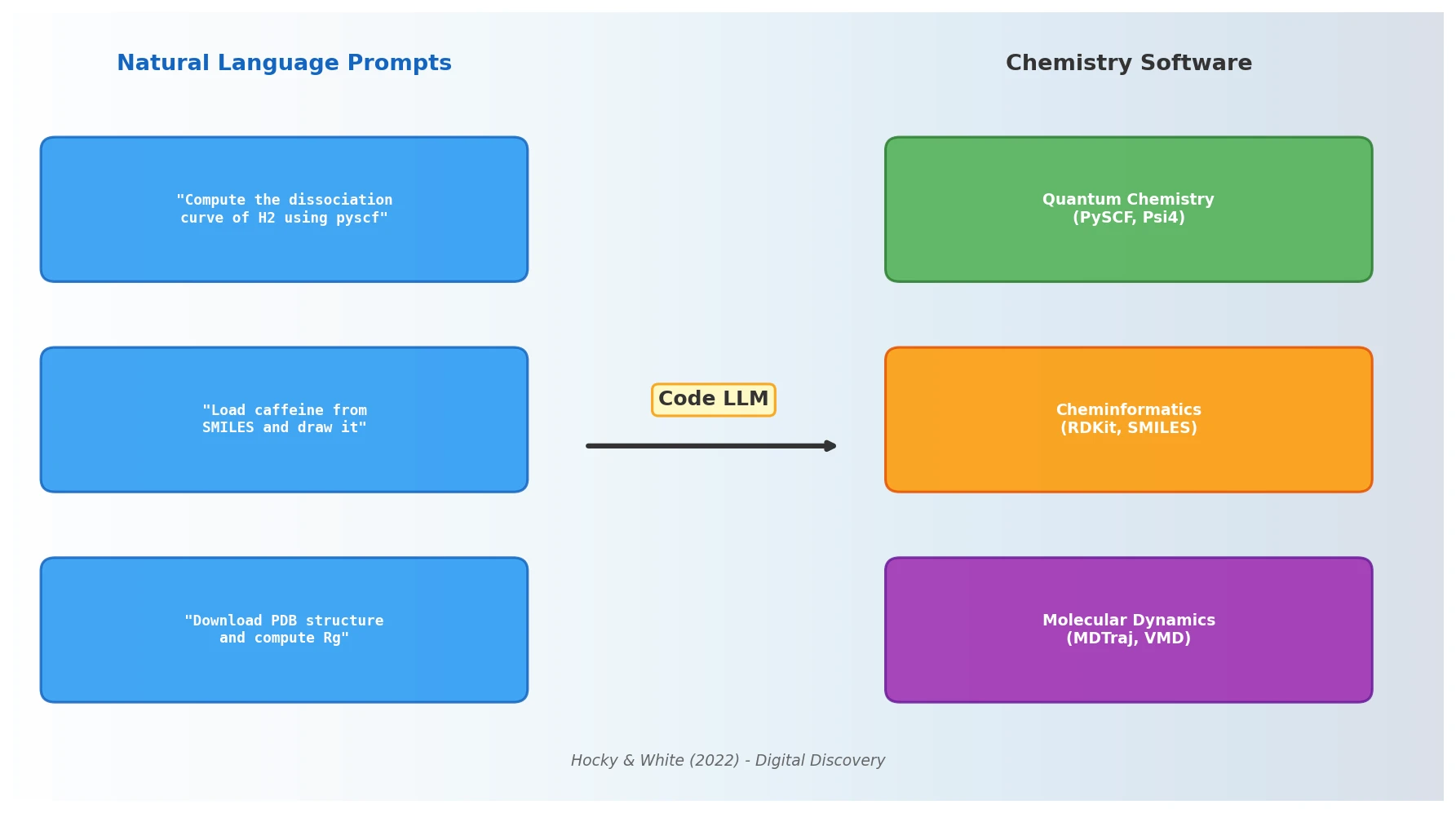
Hocky and White argue that NLP models capable of generating code from natural language prompts will fundamentally alter how chemists interact with scientific software, reducing barriers to computational research and reshaping programming pedagogy.
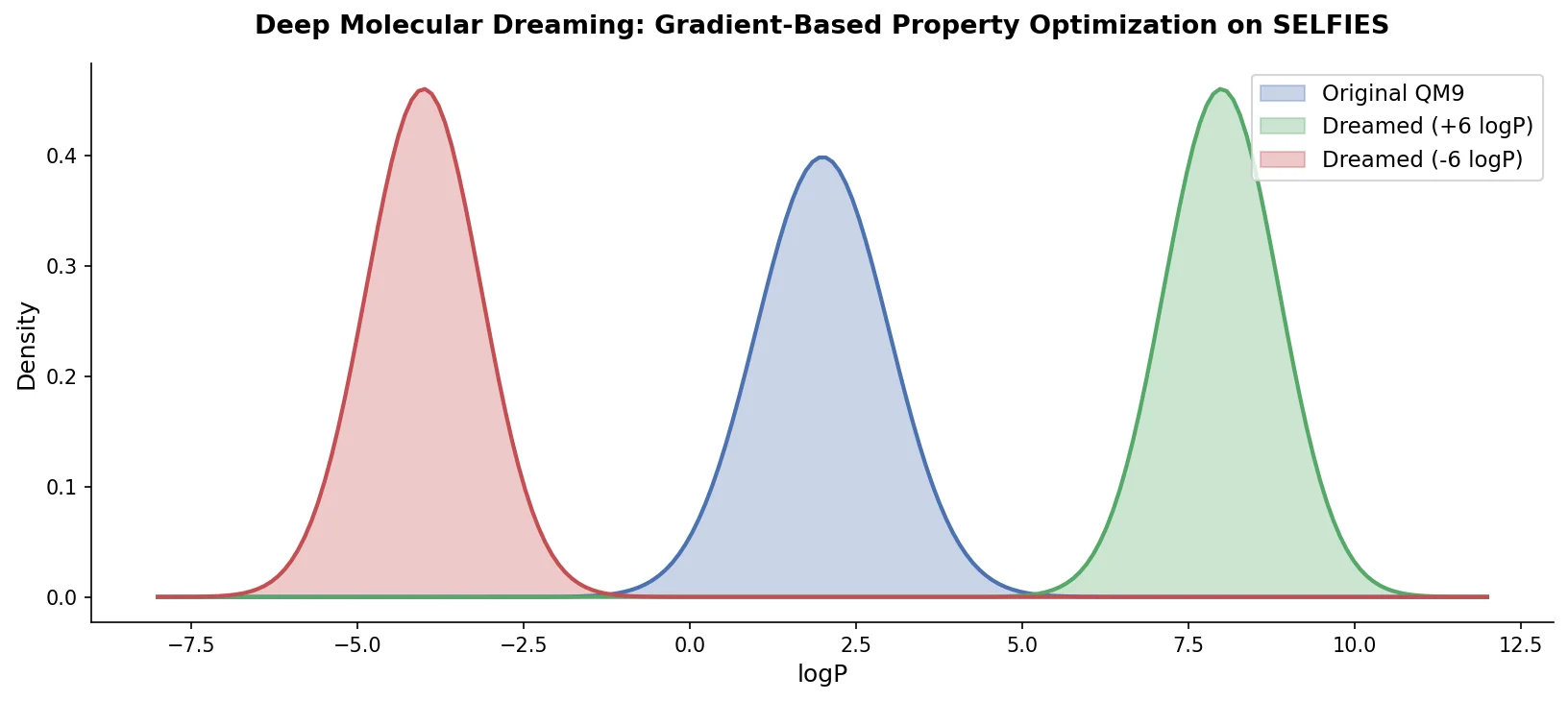
PASITHEA adapts deep dreaming from computer vision to molecular design, directly optimizing SELFIES-encoded molecules for target chemical properties via gradient-based inversion of a trained regression network.
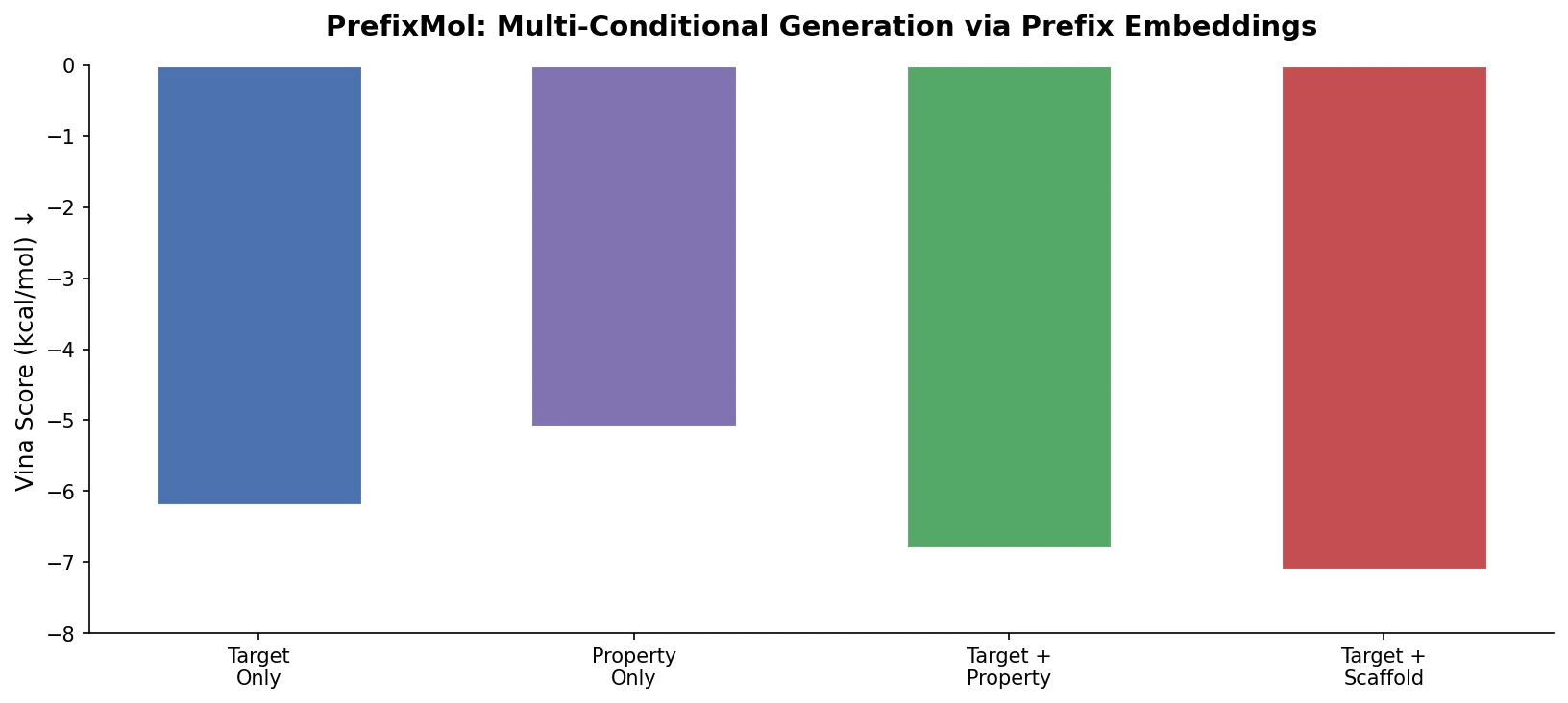
PrefixMol prepends learnable condition vectors to a GPT transformer for SMILES generation, enabling joint control over binding pocket targeting and chemical properties like QED, SA, and LogP.
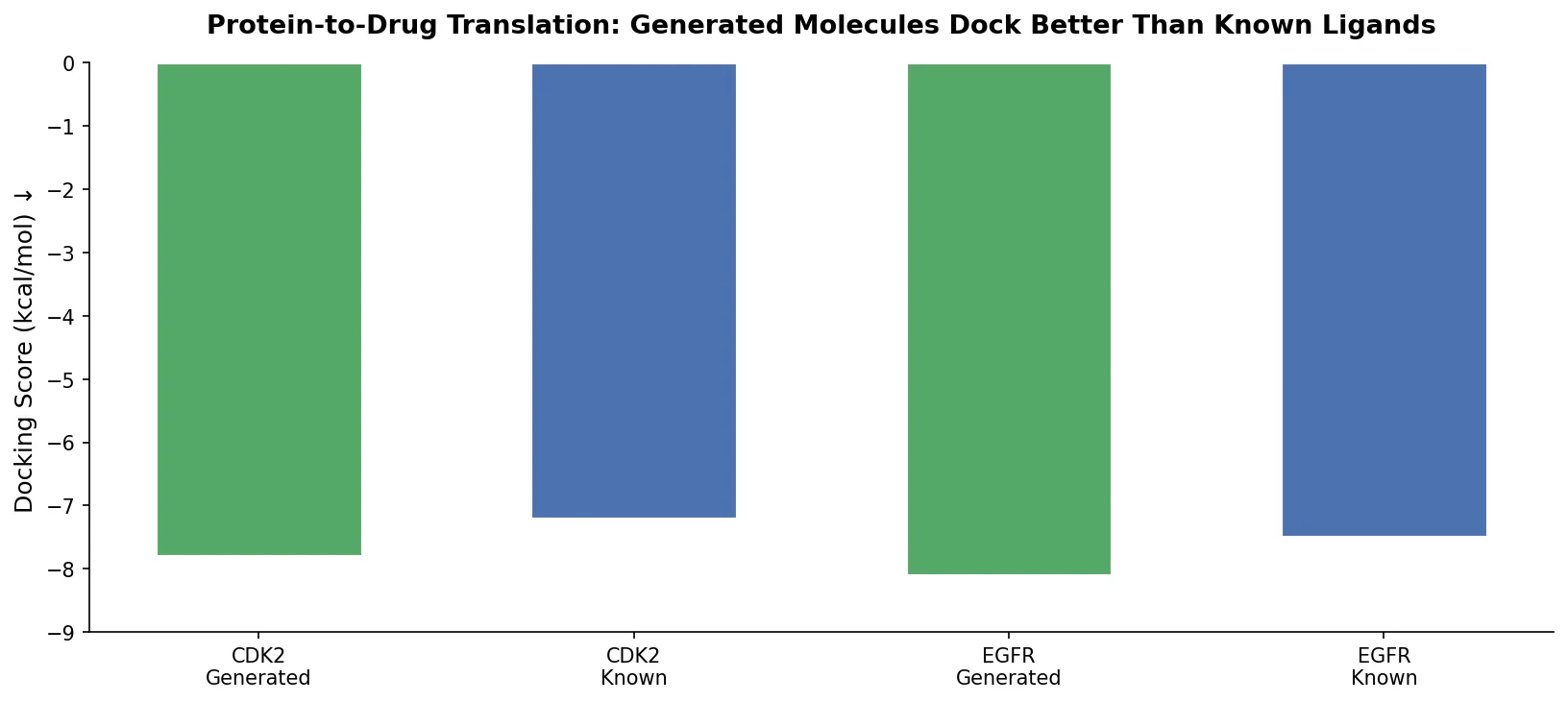
Applies the Transformer architecture to generate drug-like molecules conditioned on protein amino acid sequences, treating target-specific de novo drug design as a sequence-to-sequence translation problem.
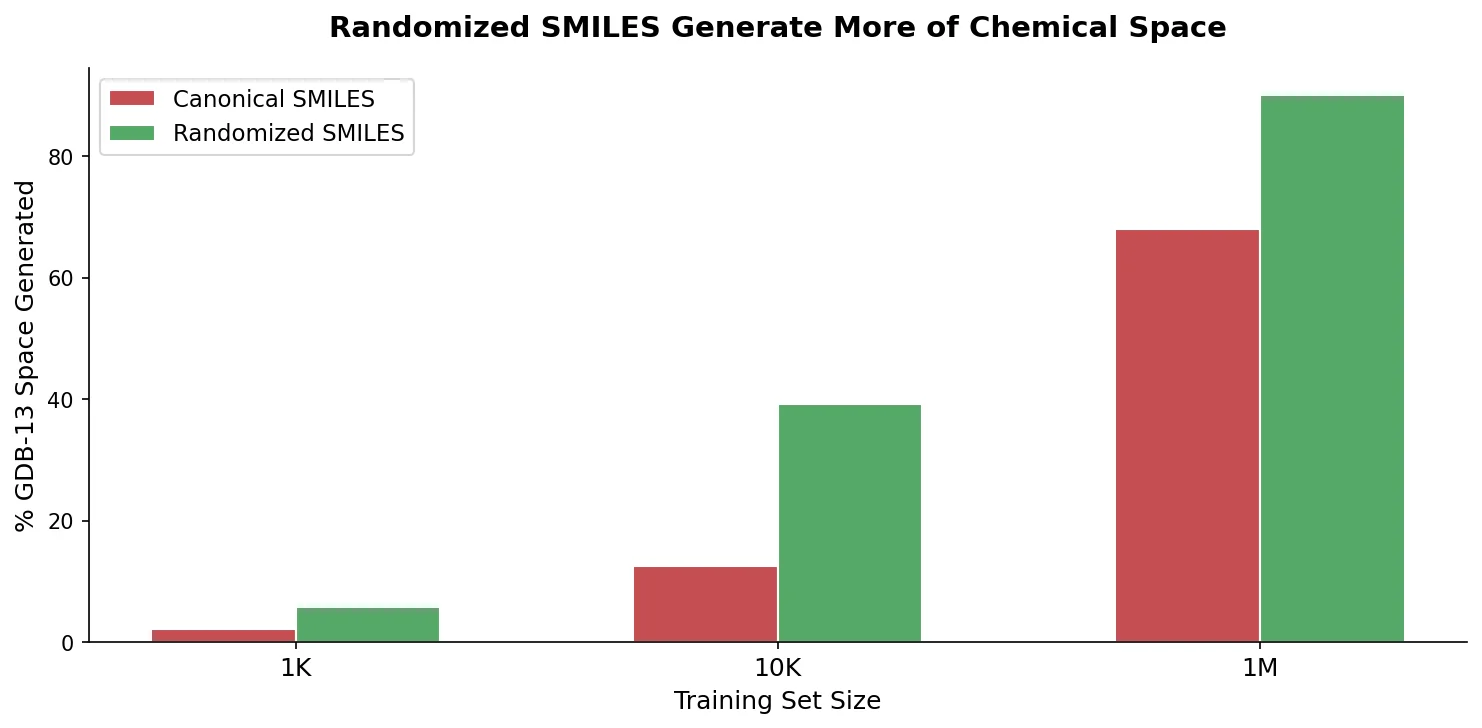
An extensive benchmark showing that training RNN generative models with randomized (non-canonical) SMILES strings yields more uniform, complete, and closed molecular output domains than canonical SMILES.
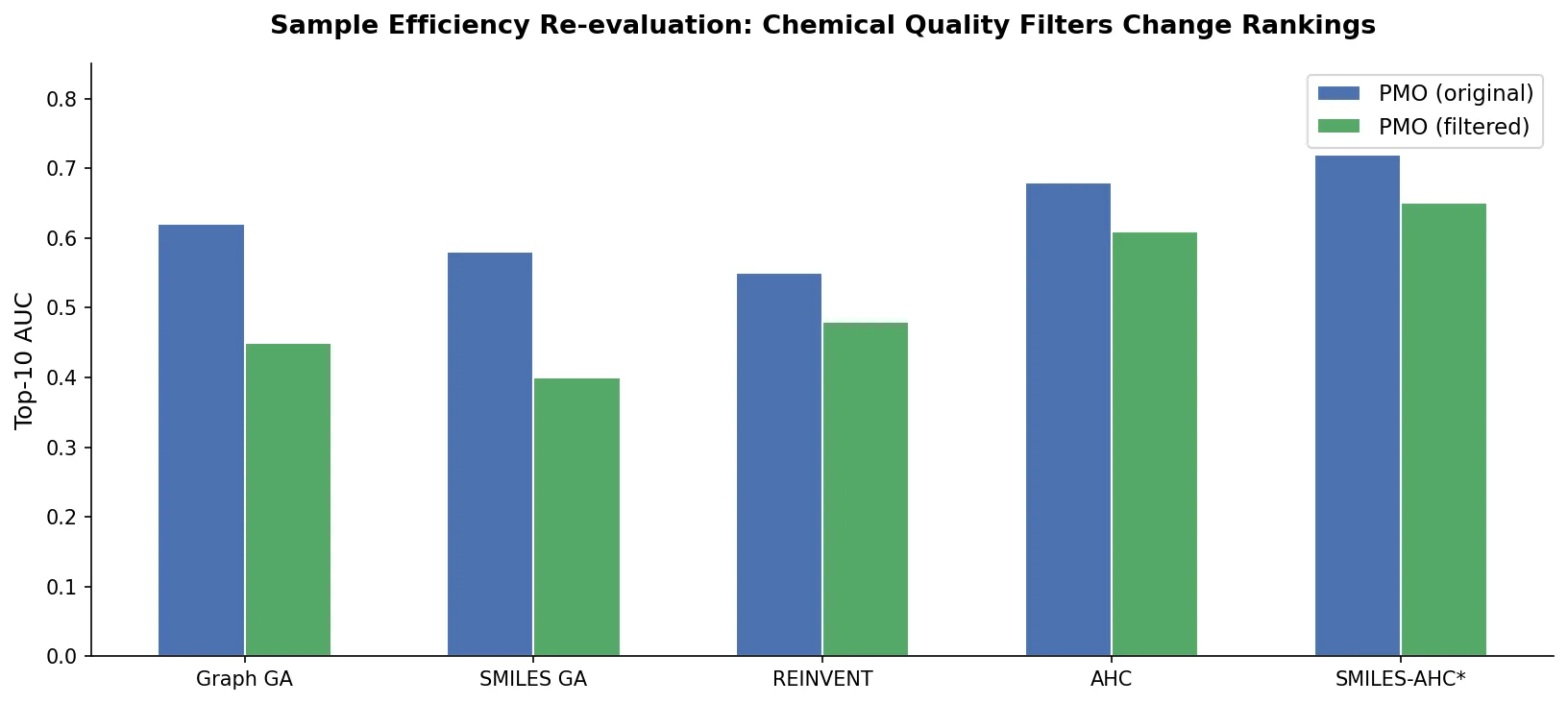
A critical reassessment of the PMO benchmark for de novo molecule generation, showing that adding molecular weight, LogP, and diversity filters substantially re-ranks generative models, with Augmented Hill-Climb emerging as the top method.
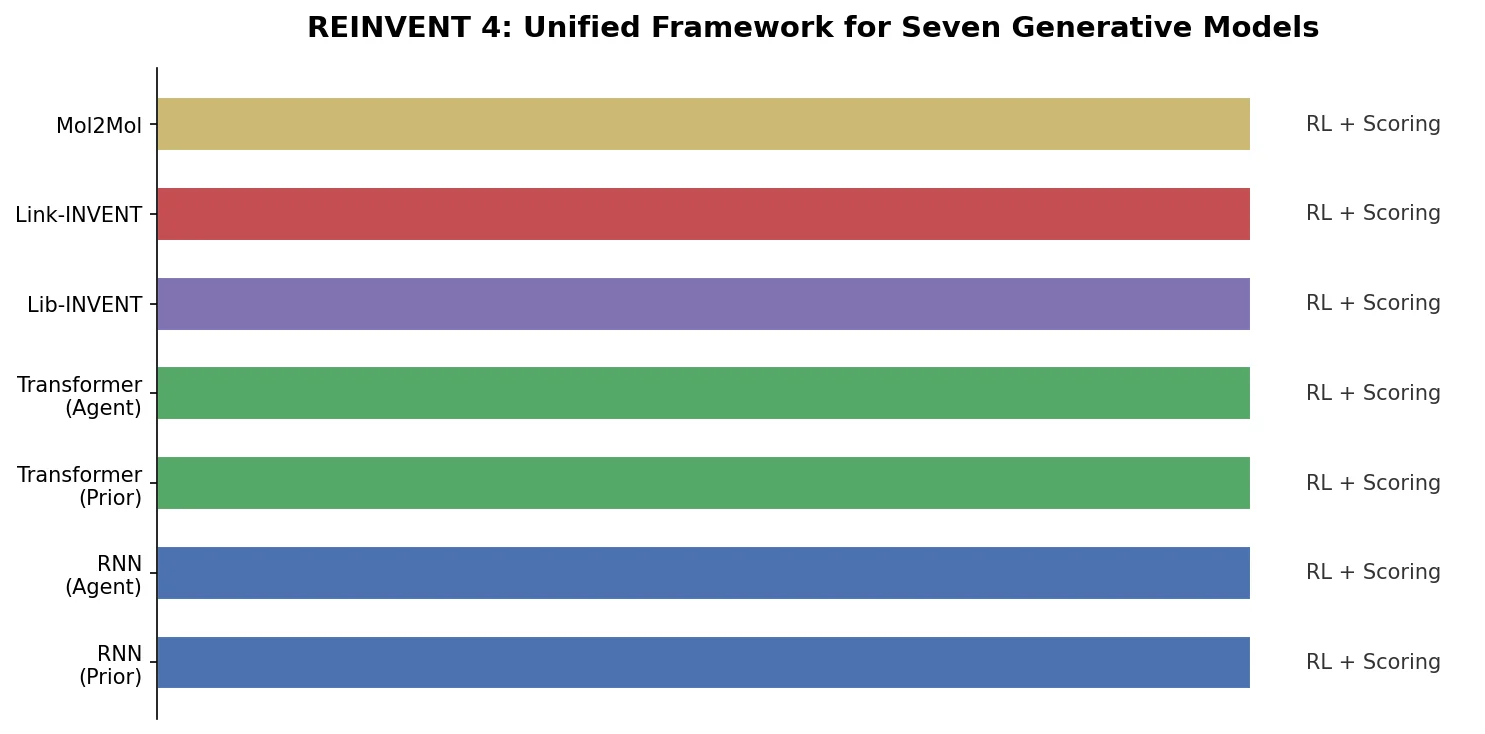
Overview of REINVENT 4, an open-source generative molecular design framework from AstraZeneca that unifies RNN and transformer generators within reinforcement learning, transfer learning, and curriculum learning optimization algorithms.
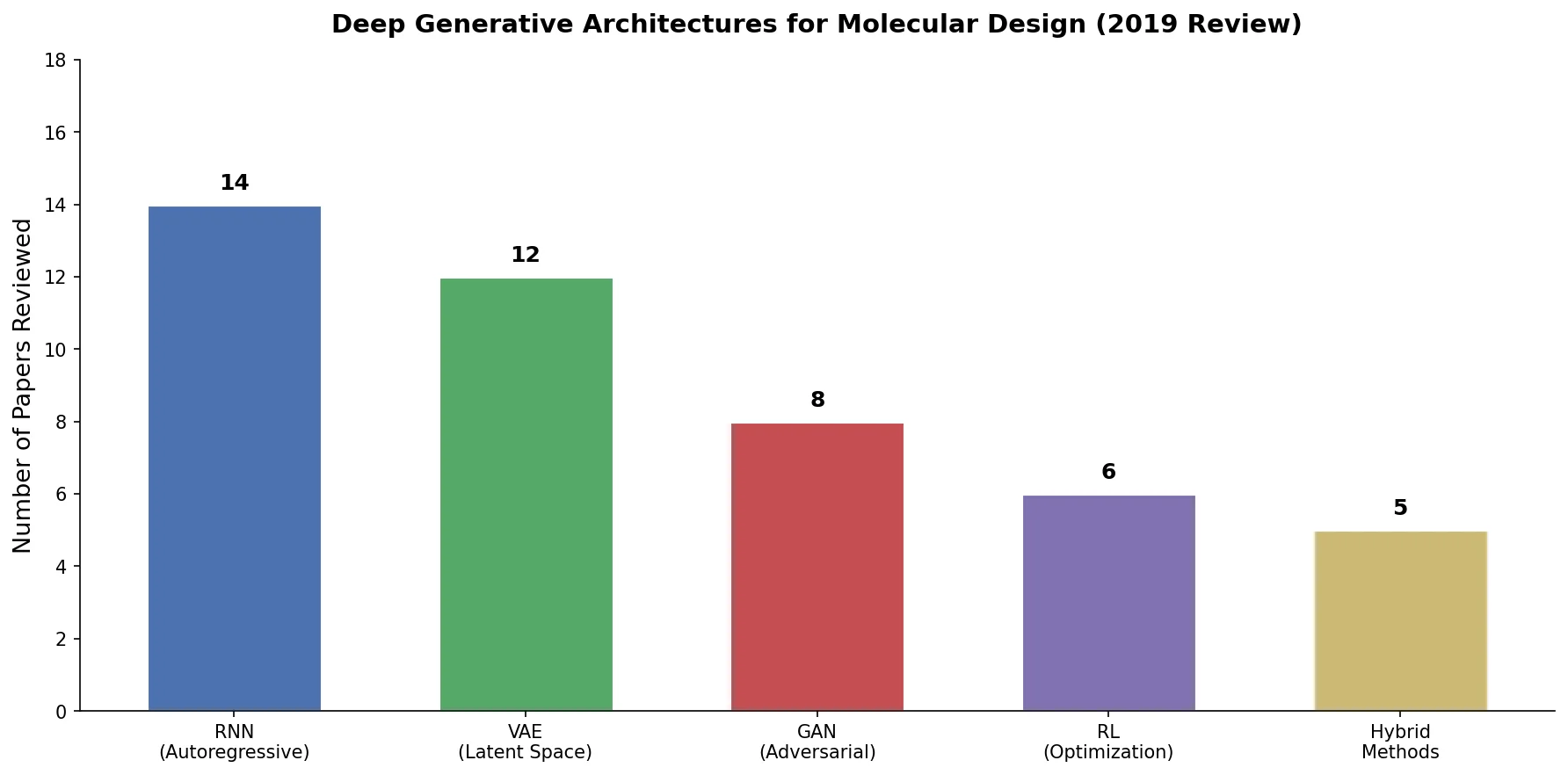
An early and influential review cataloging 45 papers on deep generative modeling for molecules, comparing RNN, VAE, GAN, and reinforcement learning architectures across SMILES and graph-based representations.
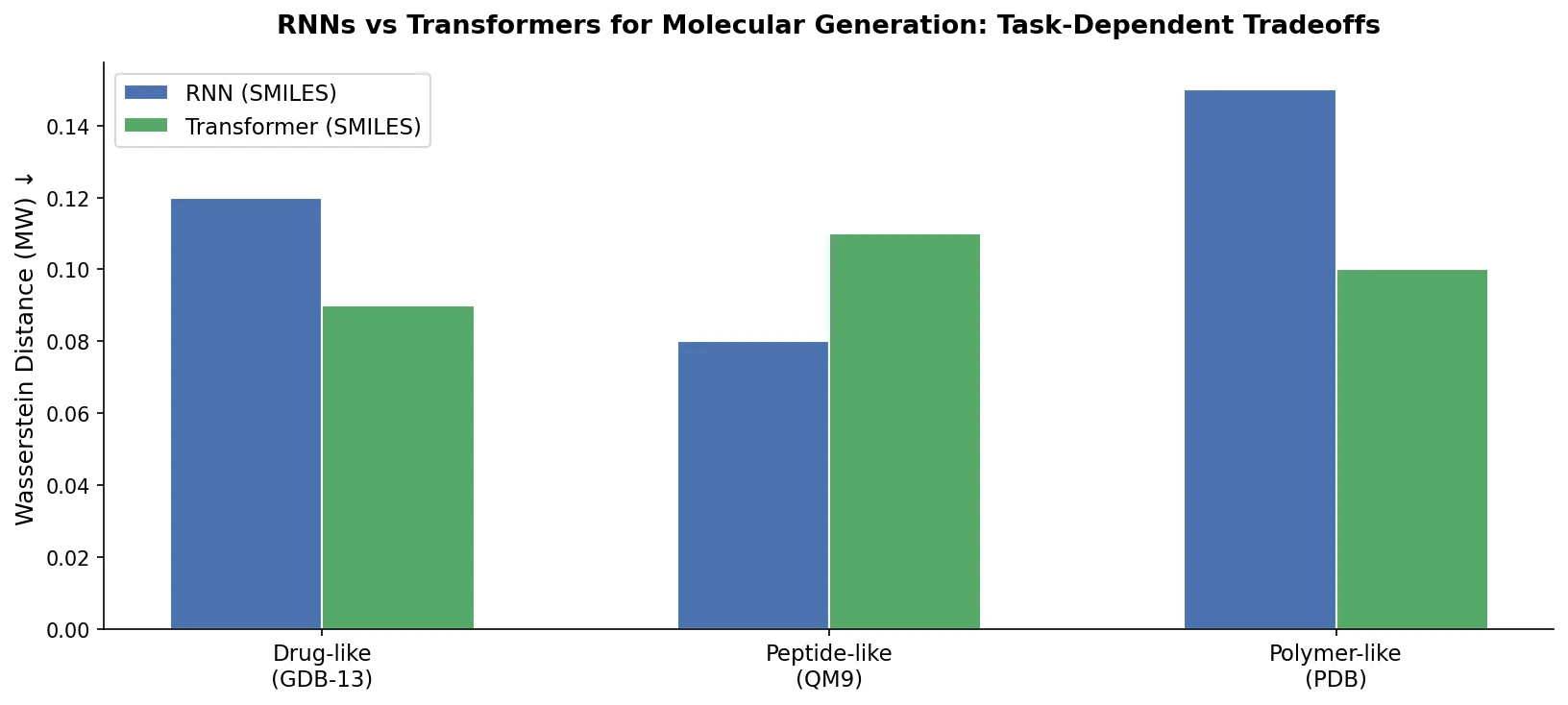
Compares RNN-based and Transformer-based chemical language models across three molecular generation tasks of increasing complexity, finding that RNNs excel at local features while Transformers handle large molecules better.
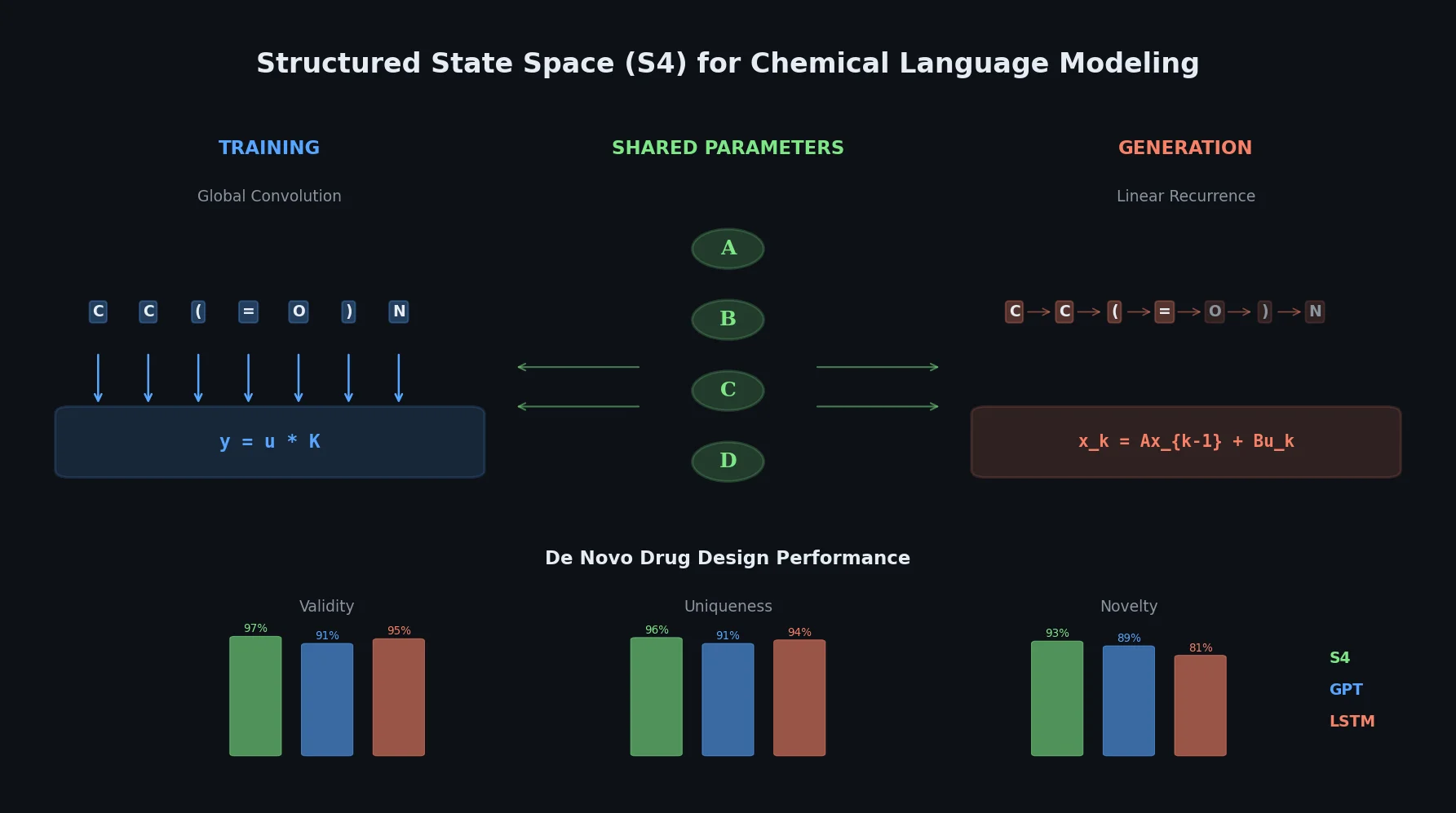
This paper introduces structured state space sequence (S4) models to chemical language modeling, showing they combine the strengths of LSTMs (efficient recurrent generation) and GPTs (holistic sequence learning) for de novo molecular design.
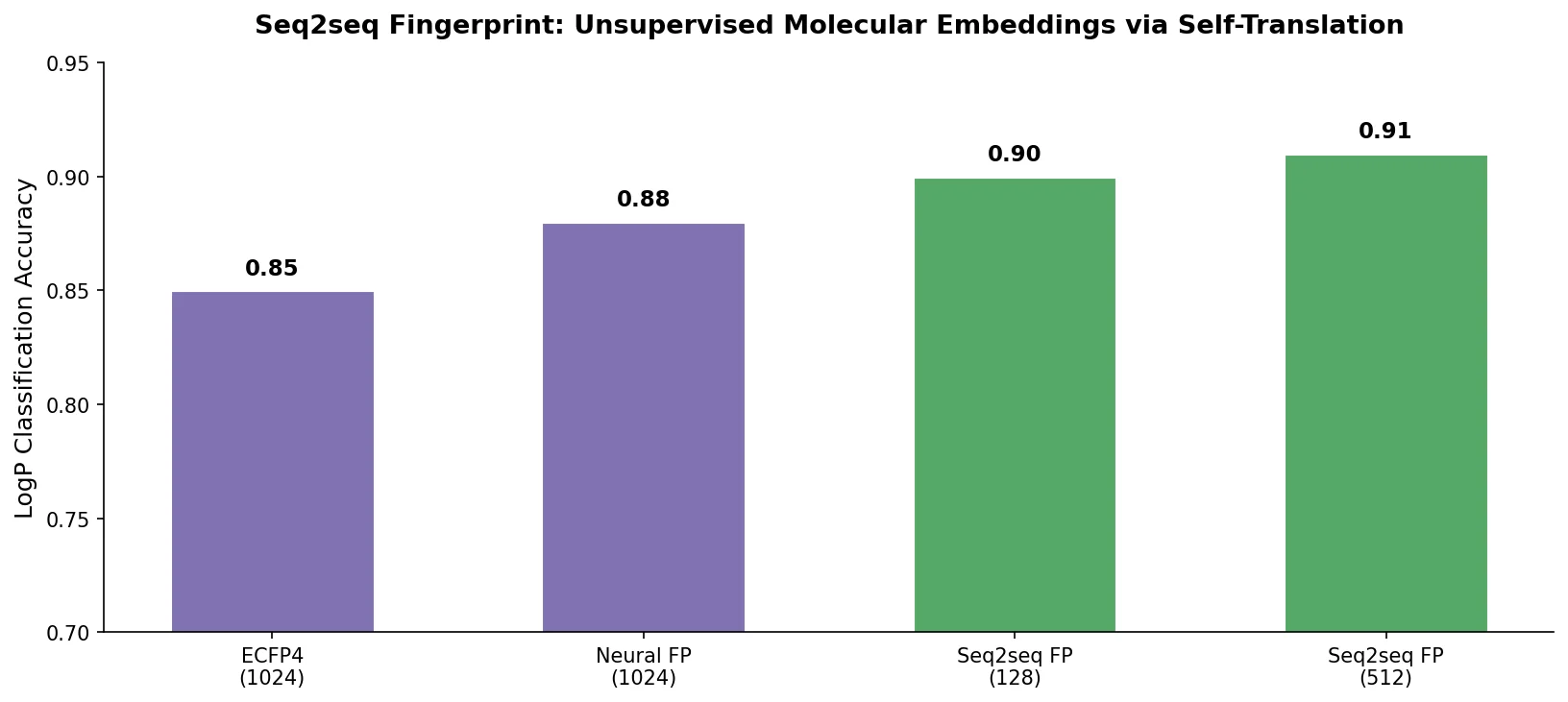
A GRU-based sequence-to-sequence model that learns fixed-length molecular fingerprints by translating SMILES strings to themselves, enabling unsupervised representation learning for drug discovery tasks.