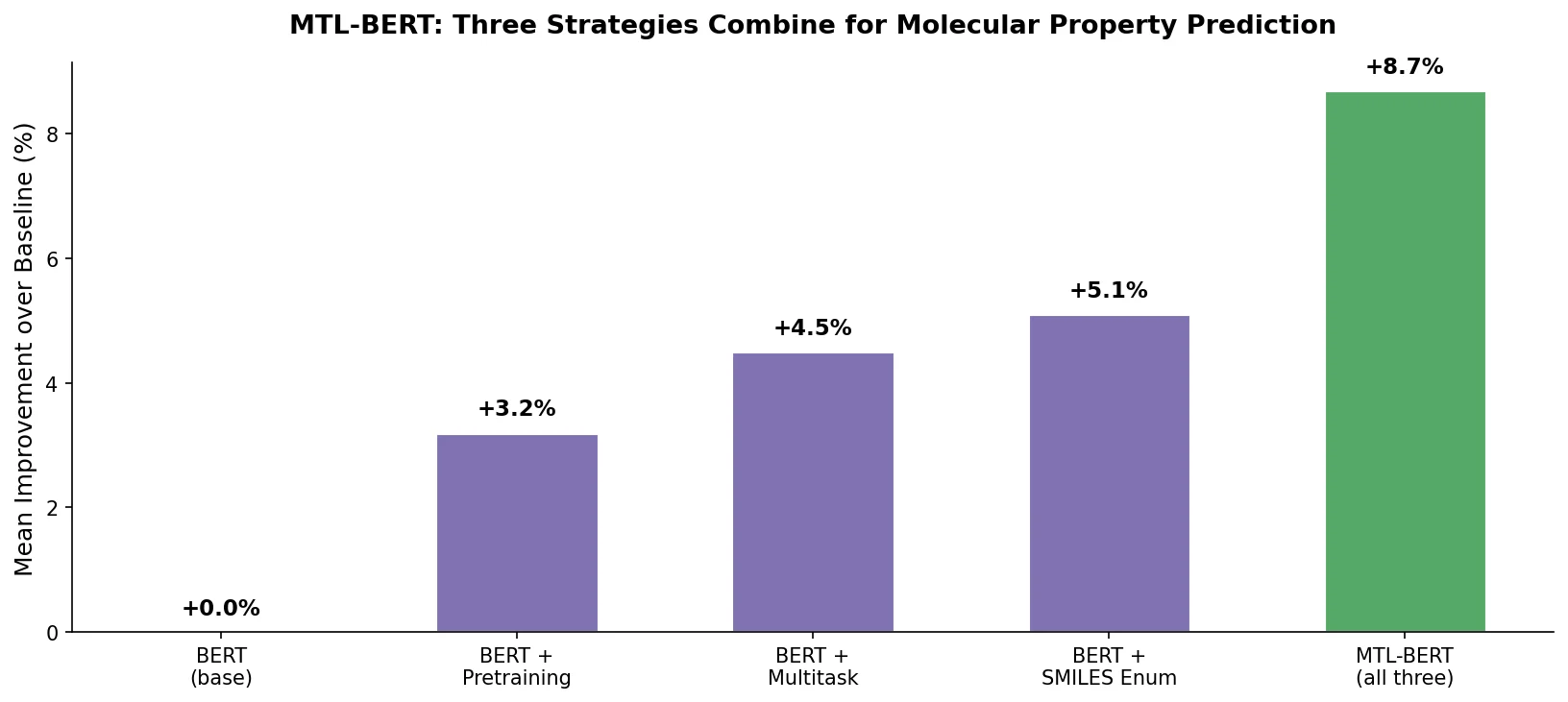
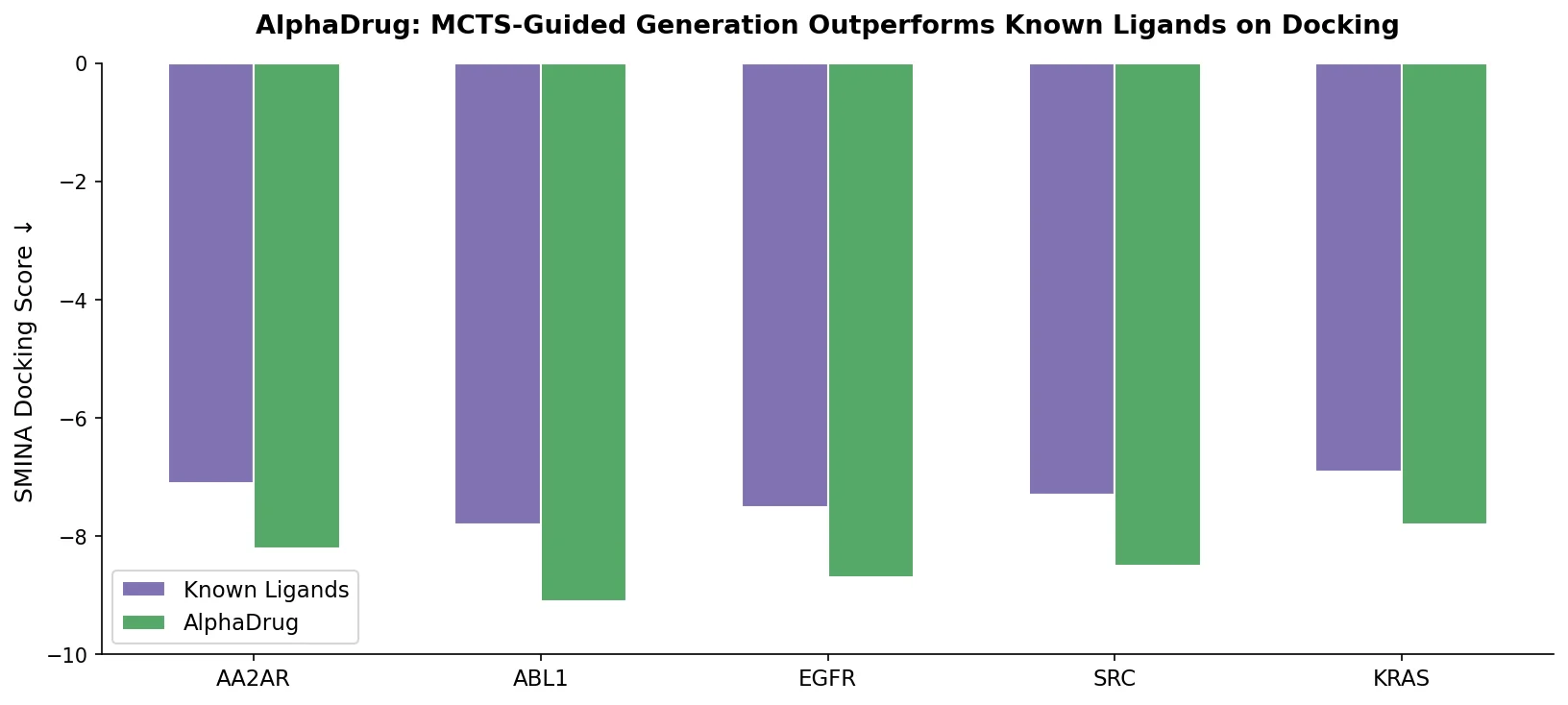
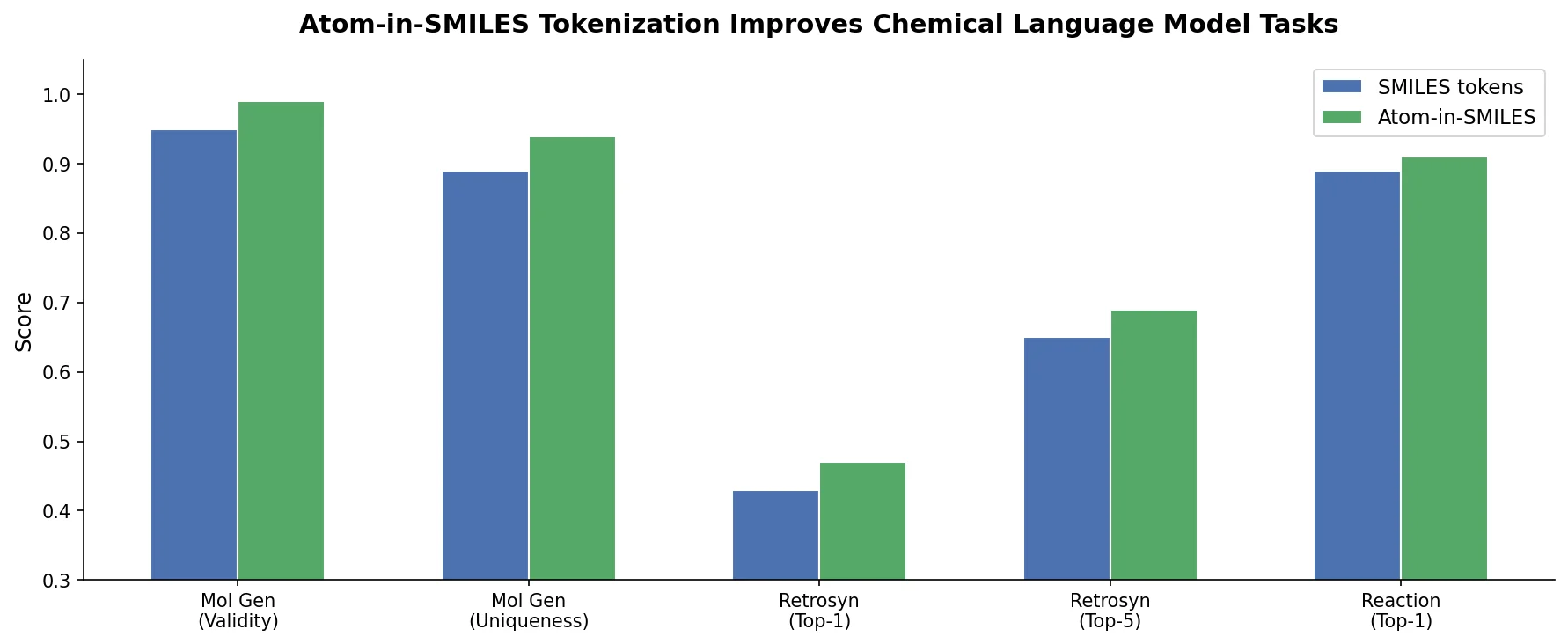
PharmaGPT: Domain-Specific LLMs for Pharma and Chem
PharmaGPT is a suite of domain-specific LLMs (13B and 70B parameters) built on LLaMA with continued pretraining on biopharmaceutical and chemical data, achieving strong results on NAPLEX and Chinese pharmacist exams.