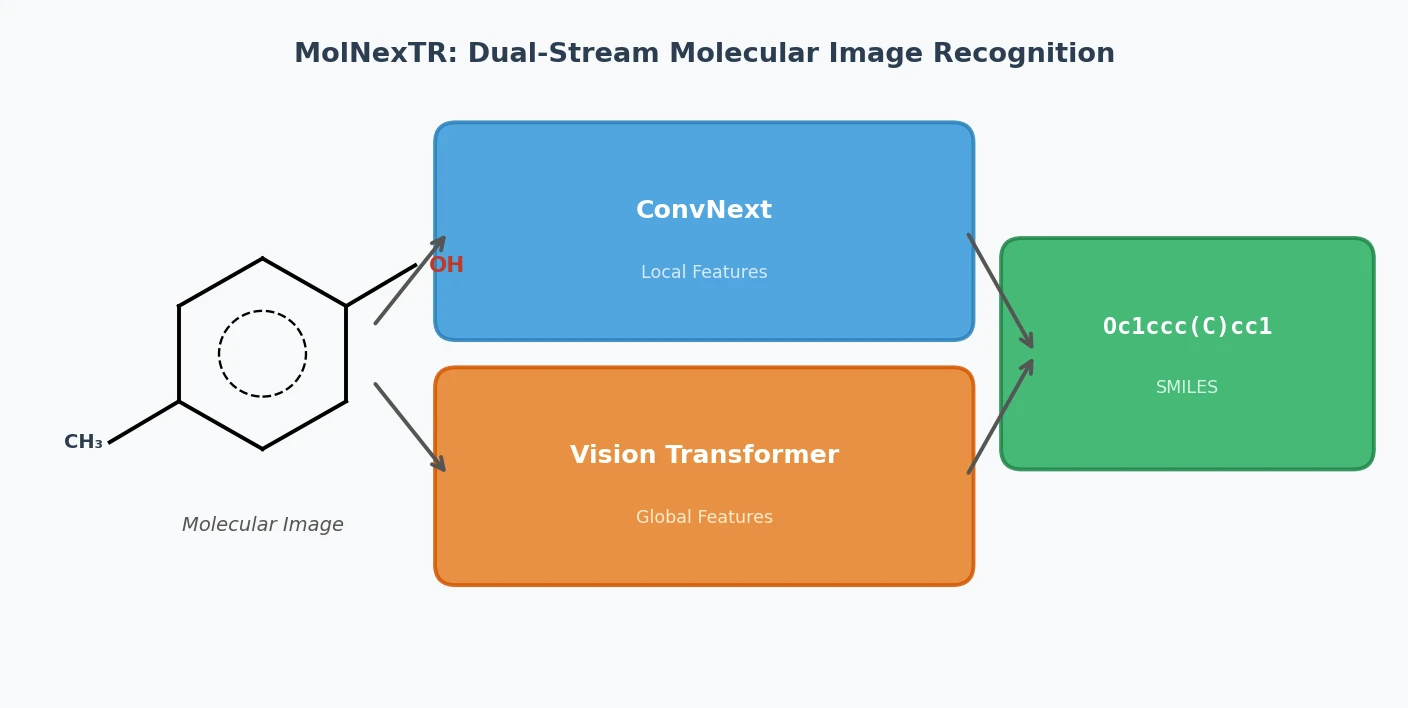
MolNexTR: A Dual-Stream Molecular Image Recognition
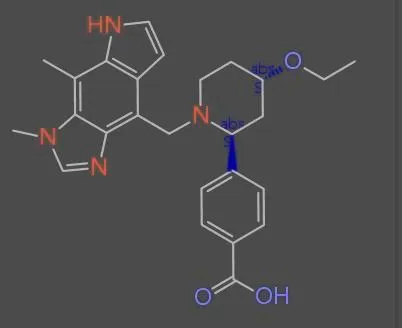
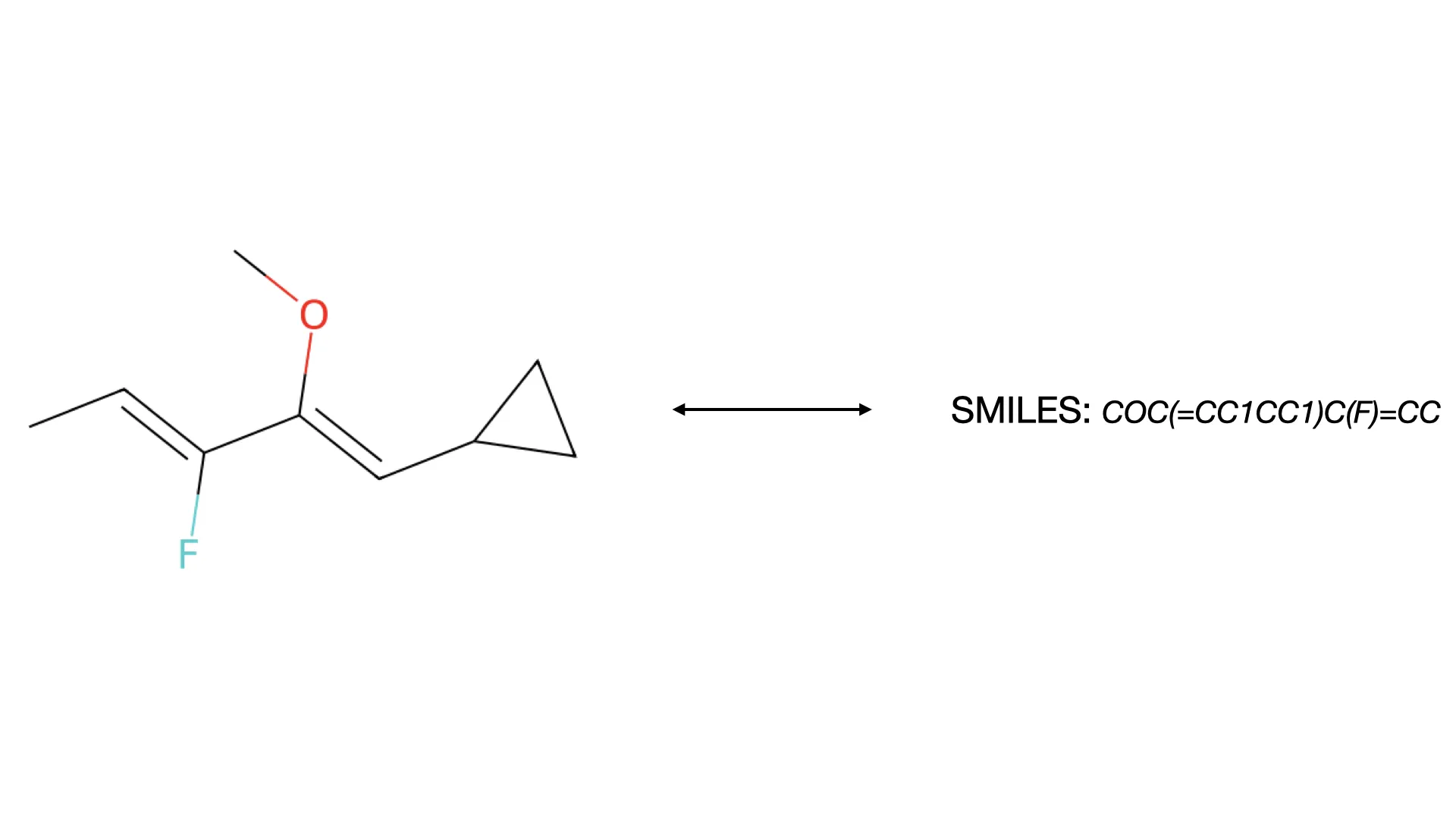
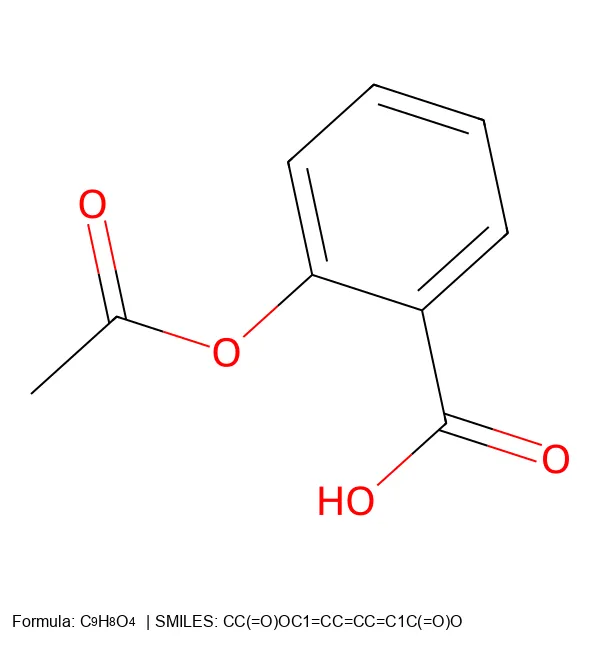
MolNexTR proposes a dual-stream architecture combining ConvNext and Vision Transformers to improve molecular image recognition (OCSR). It achieves 81-97% accuracy across diverse benchmarks utilizing simultaneous local and global feature extraction alongside specialized image contamination augmentations.