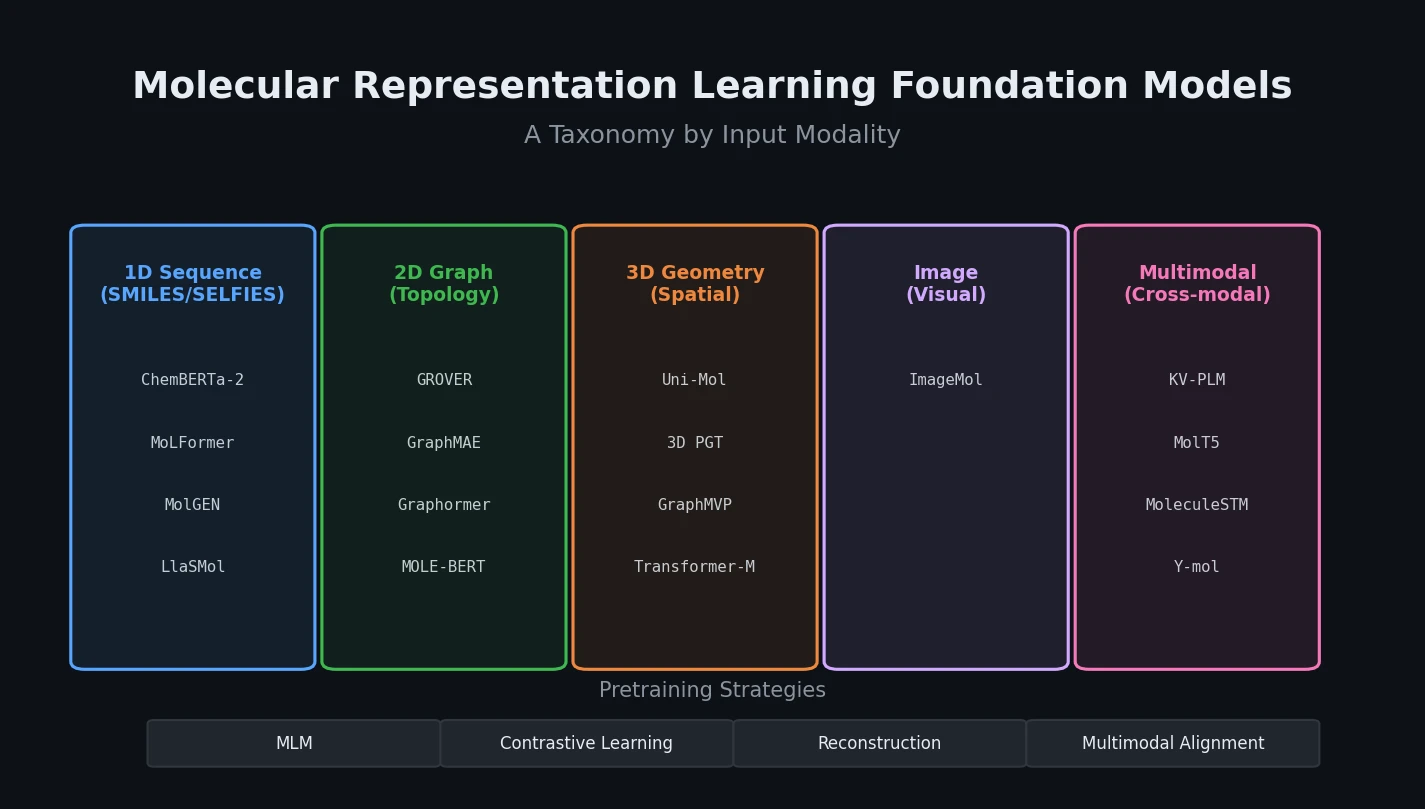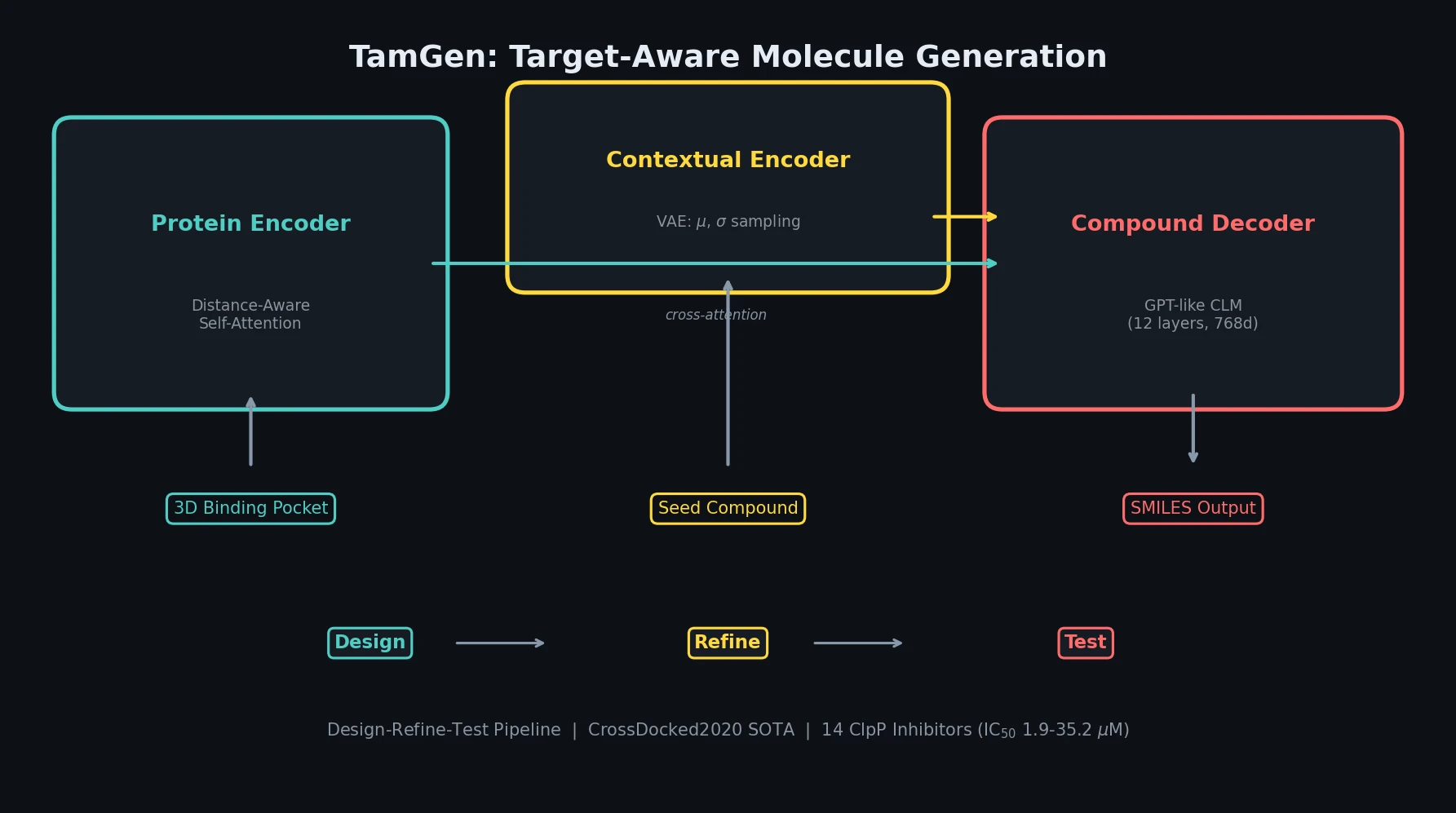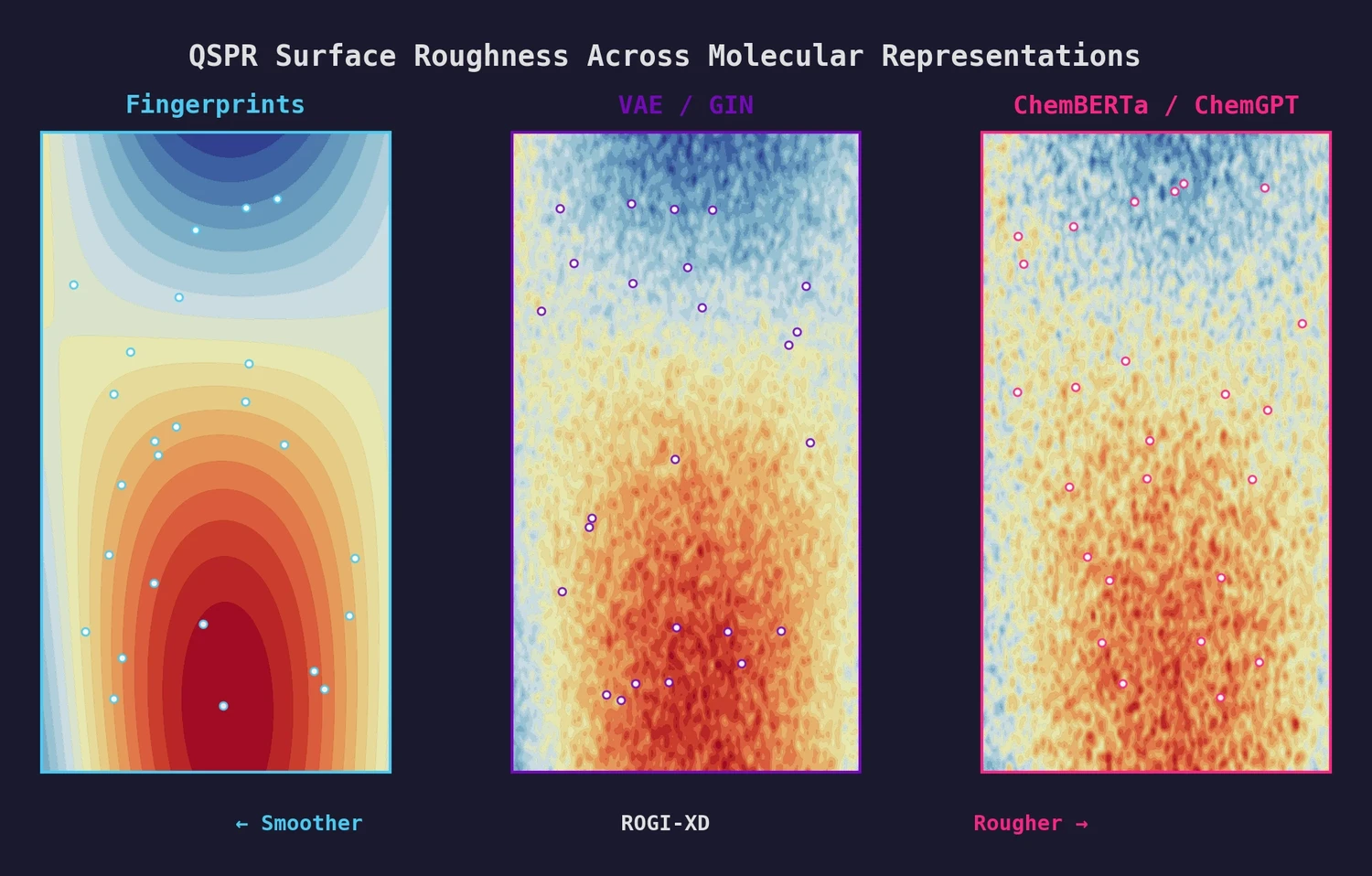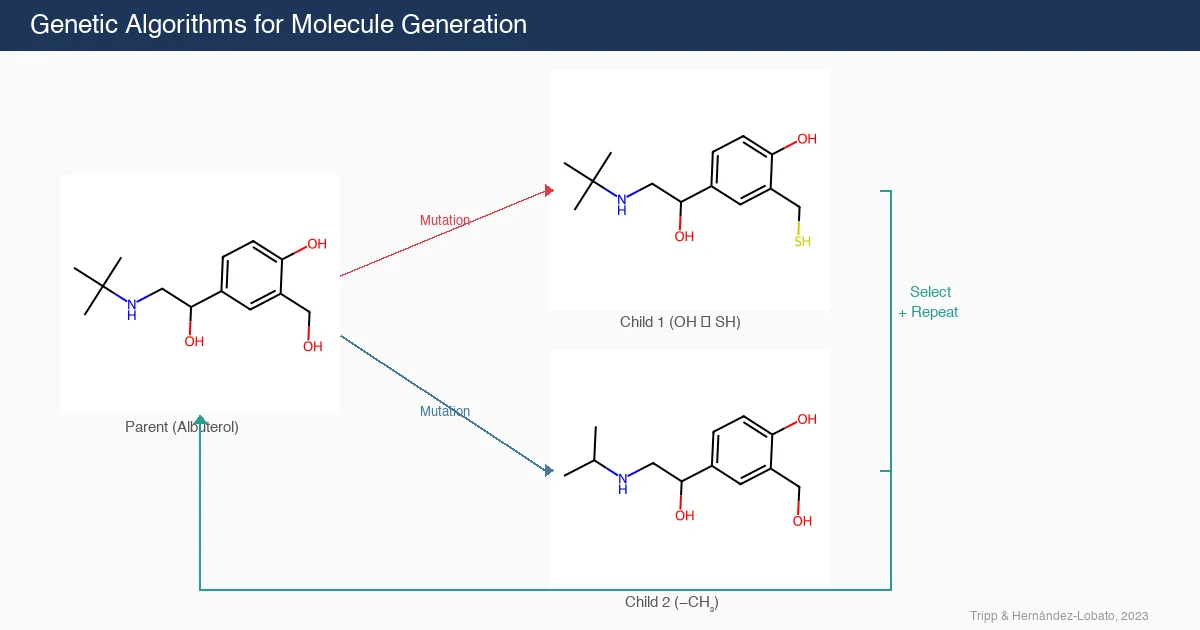
PMO: Benchmarking Sample-Efficient Molecular Design
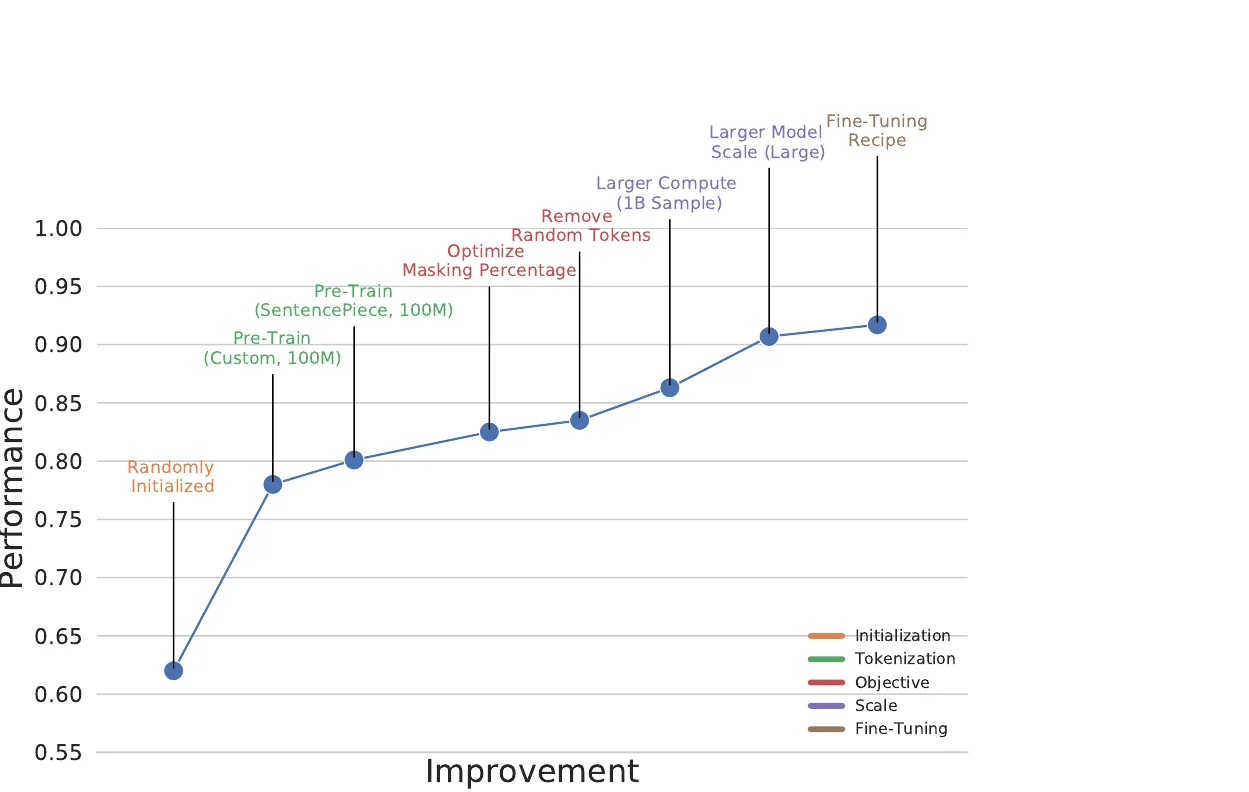
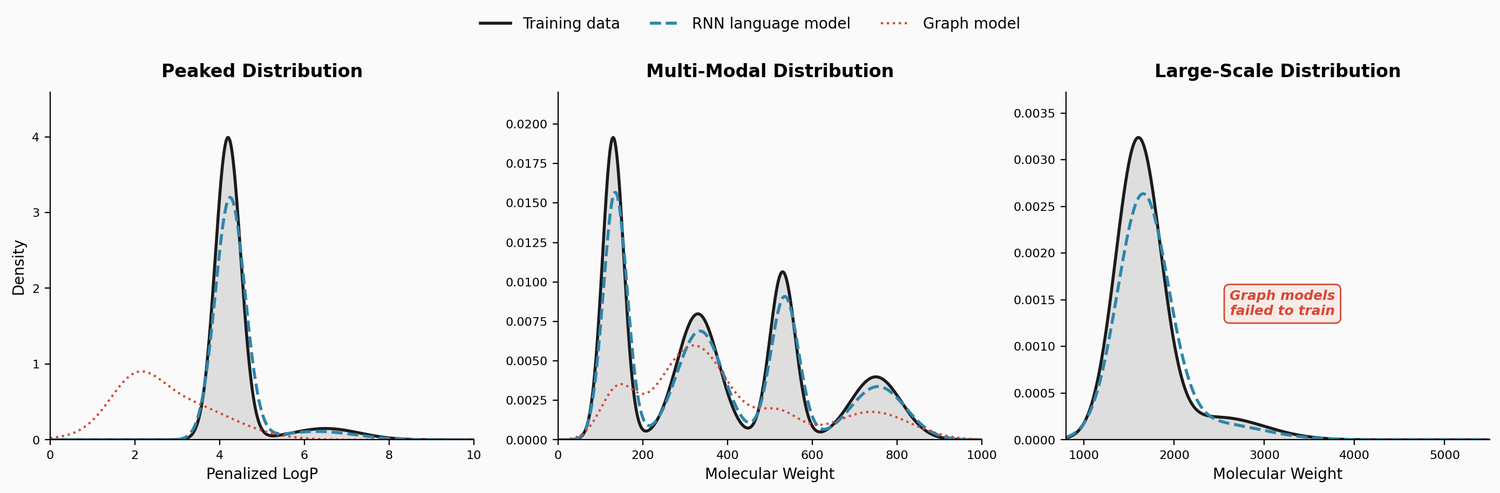
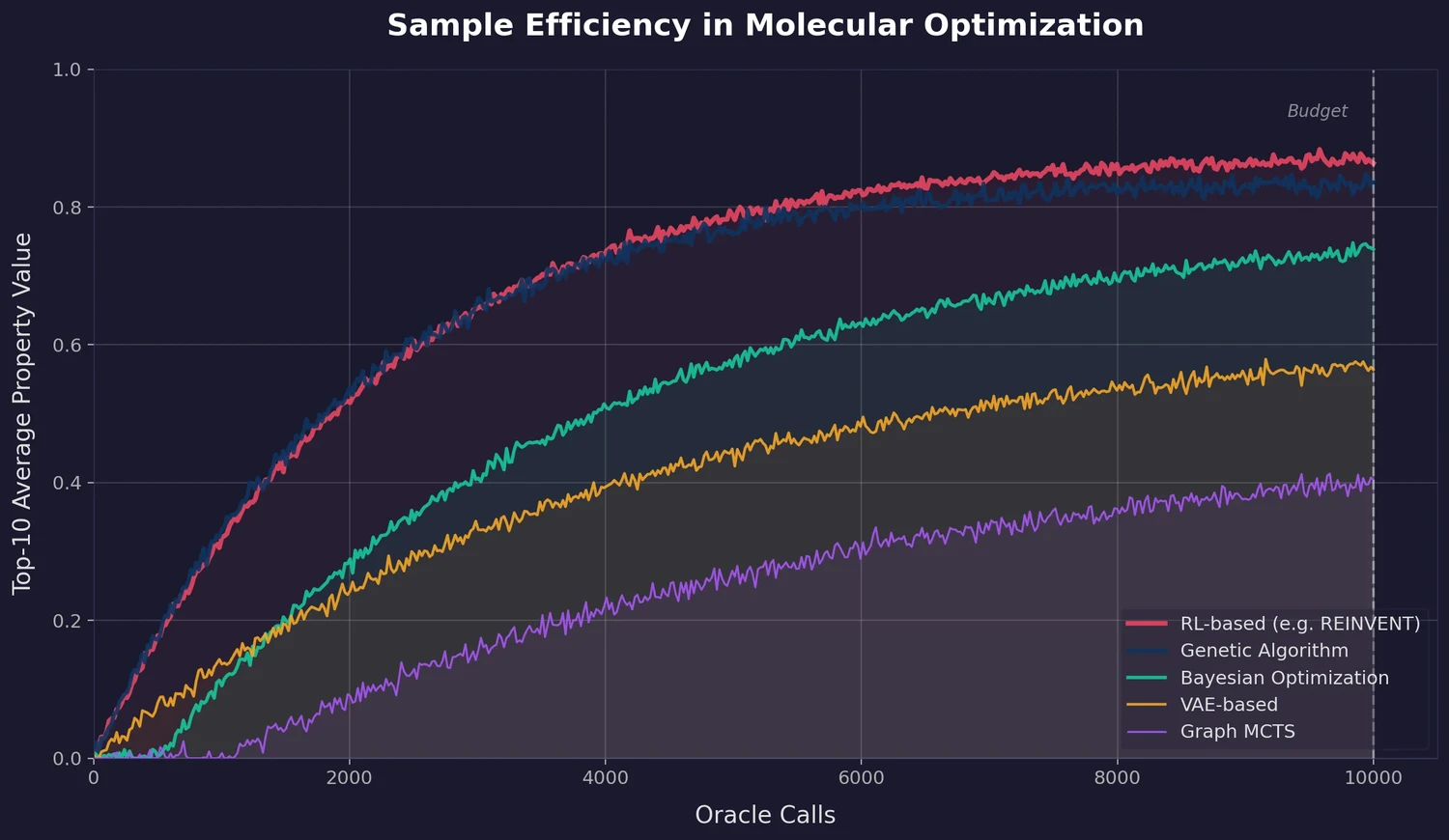
A large-scale benchmark of 25 molecular optimization methods on 23 oracles under constrained oracle budgets, showing that sample efficiency is a critical and often neglected dimension of evaluation.