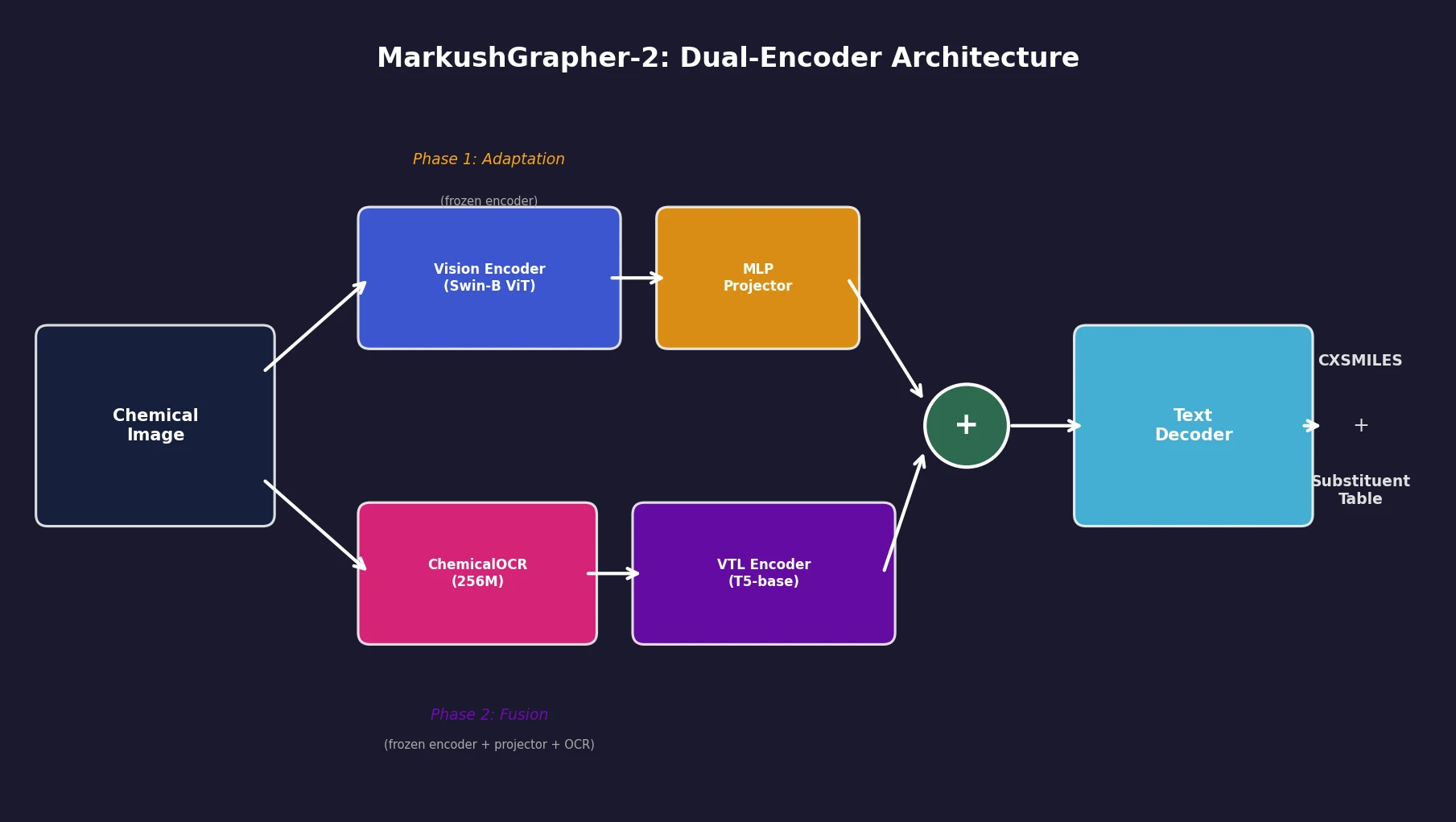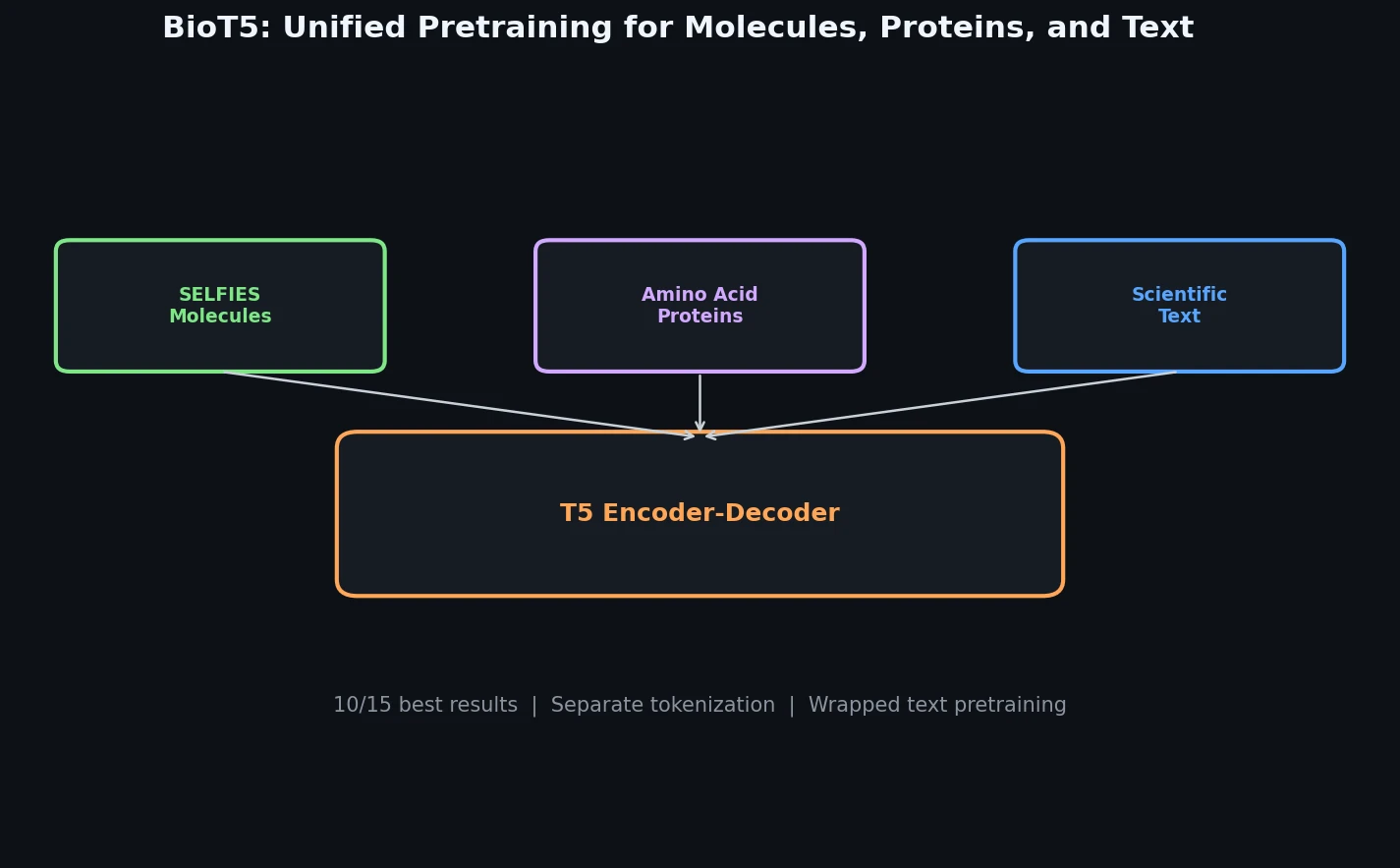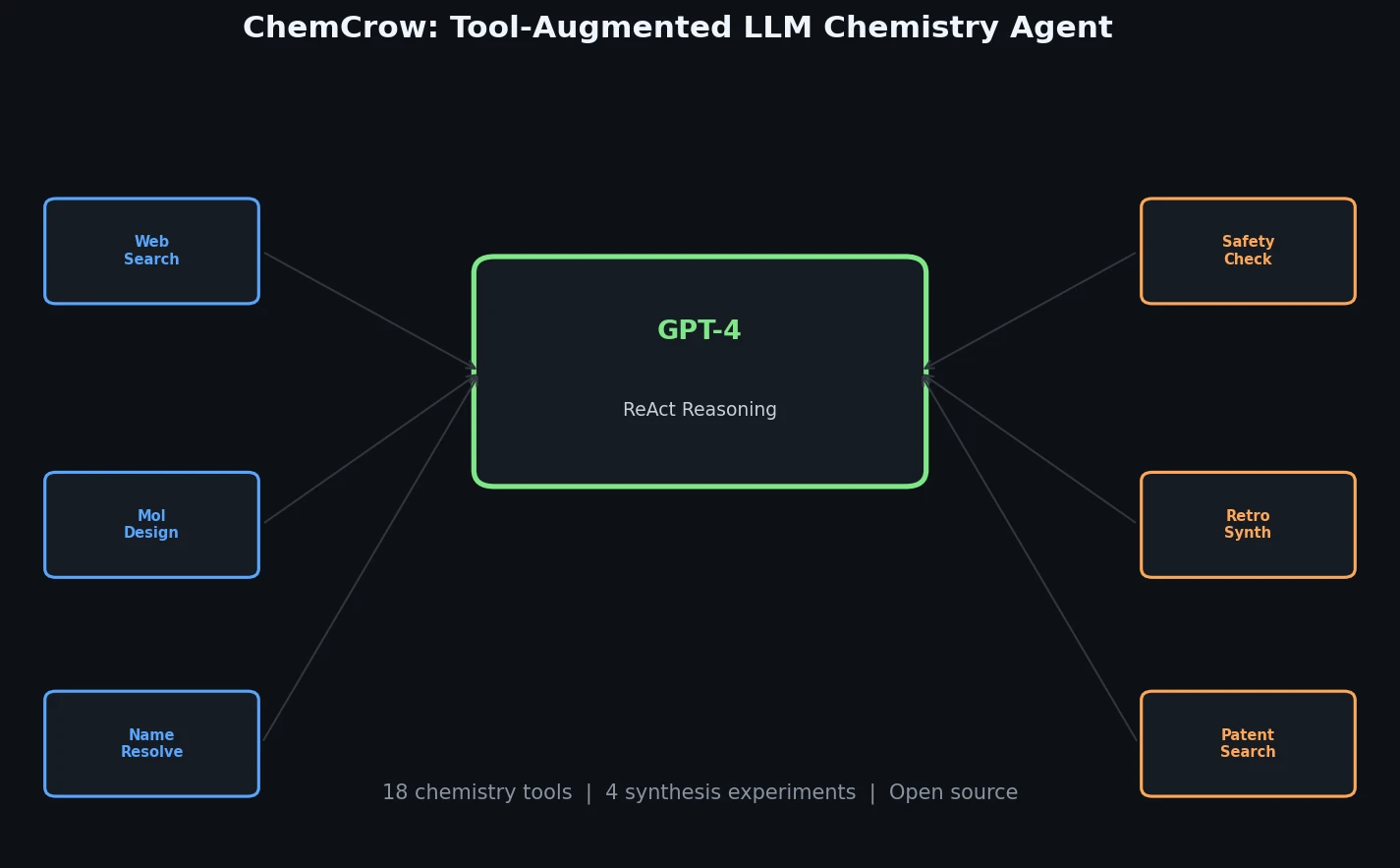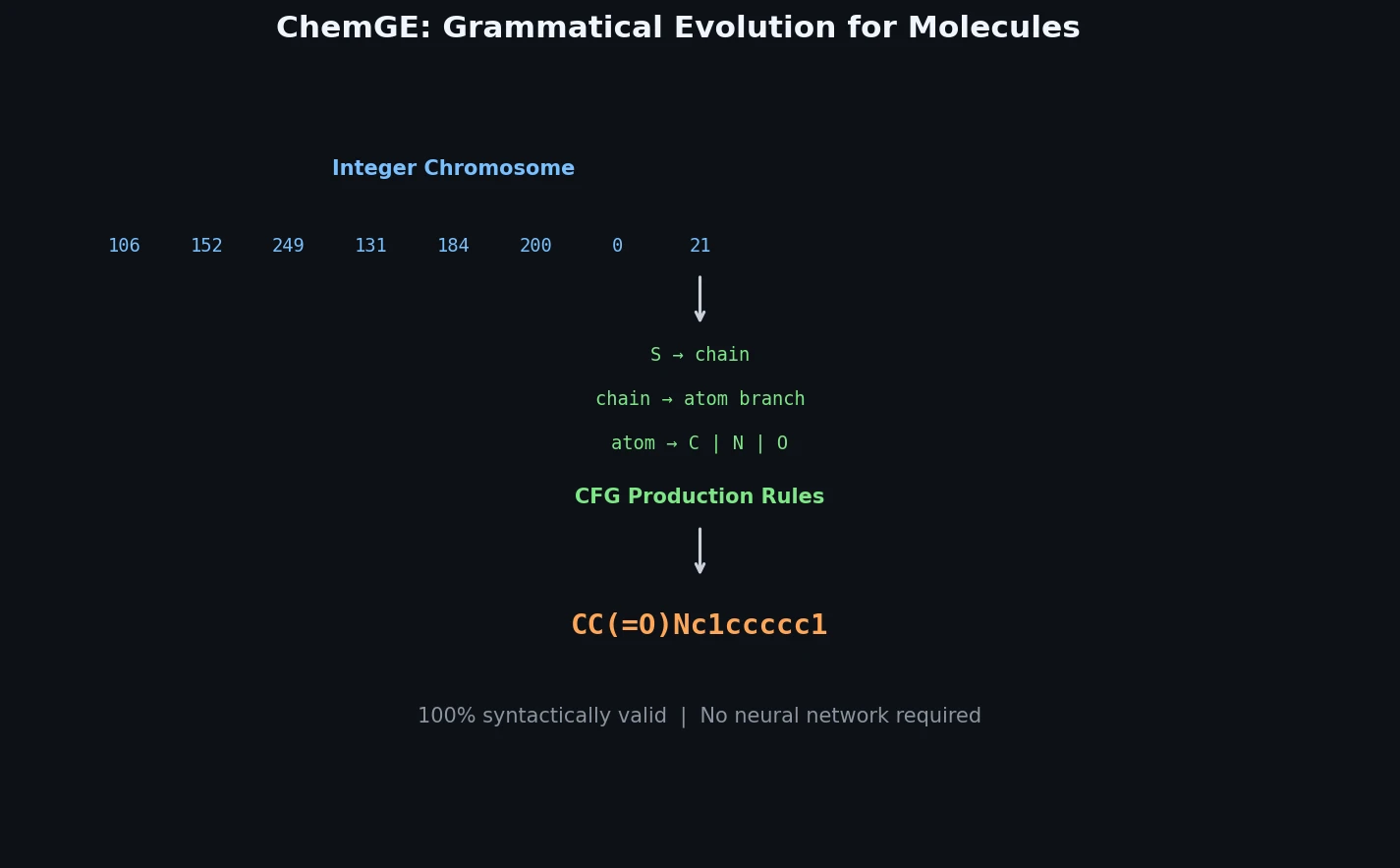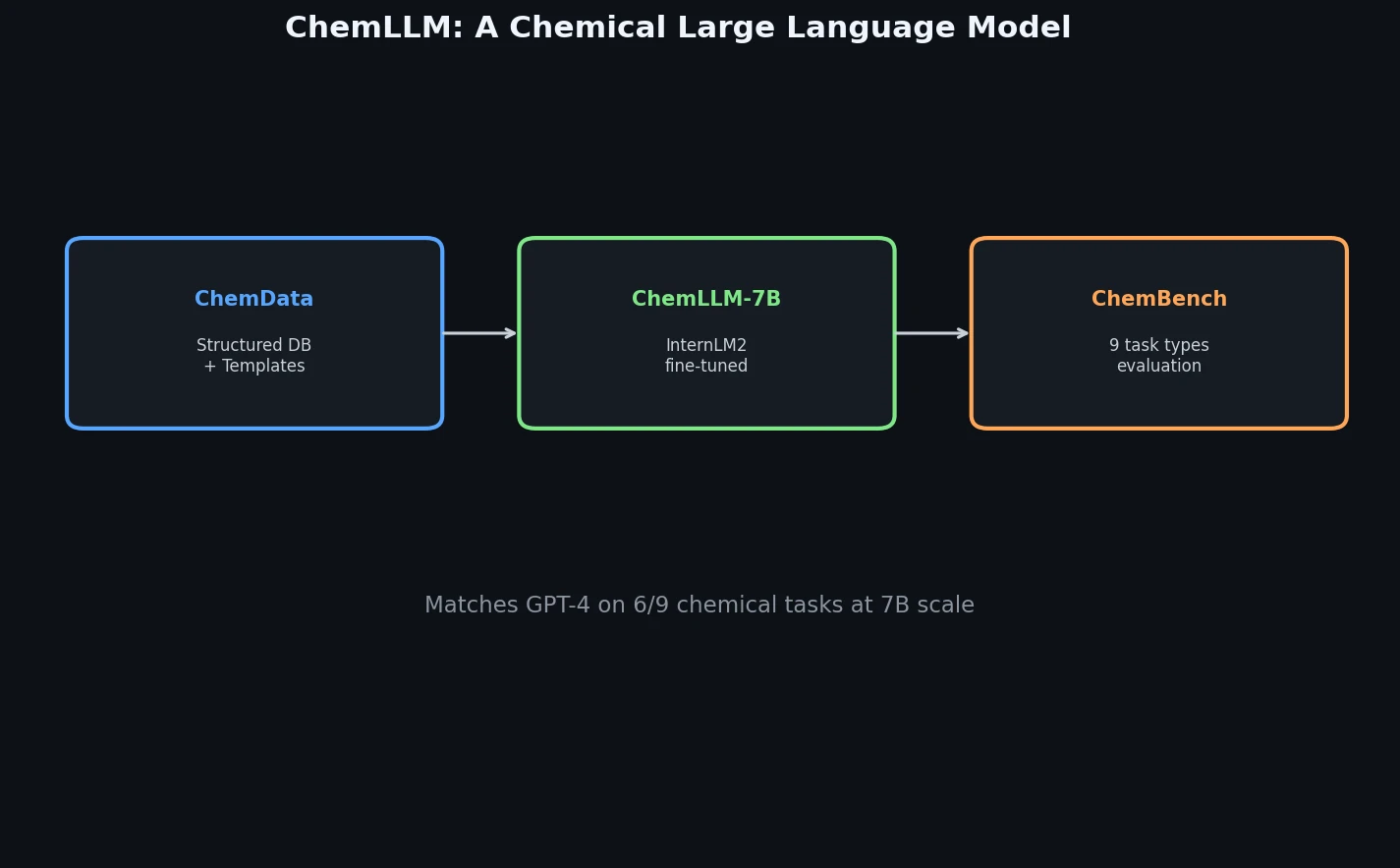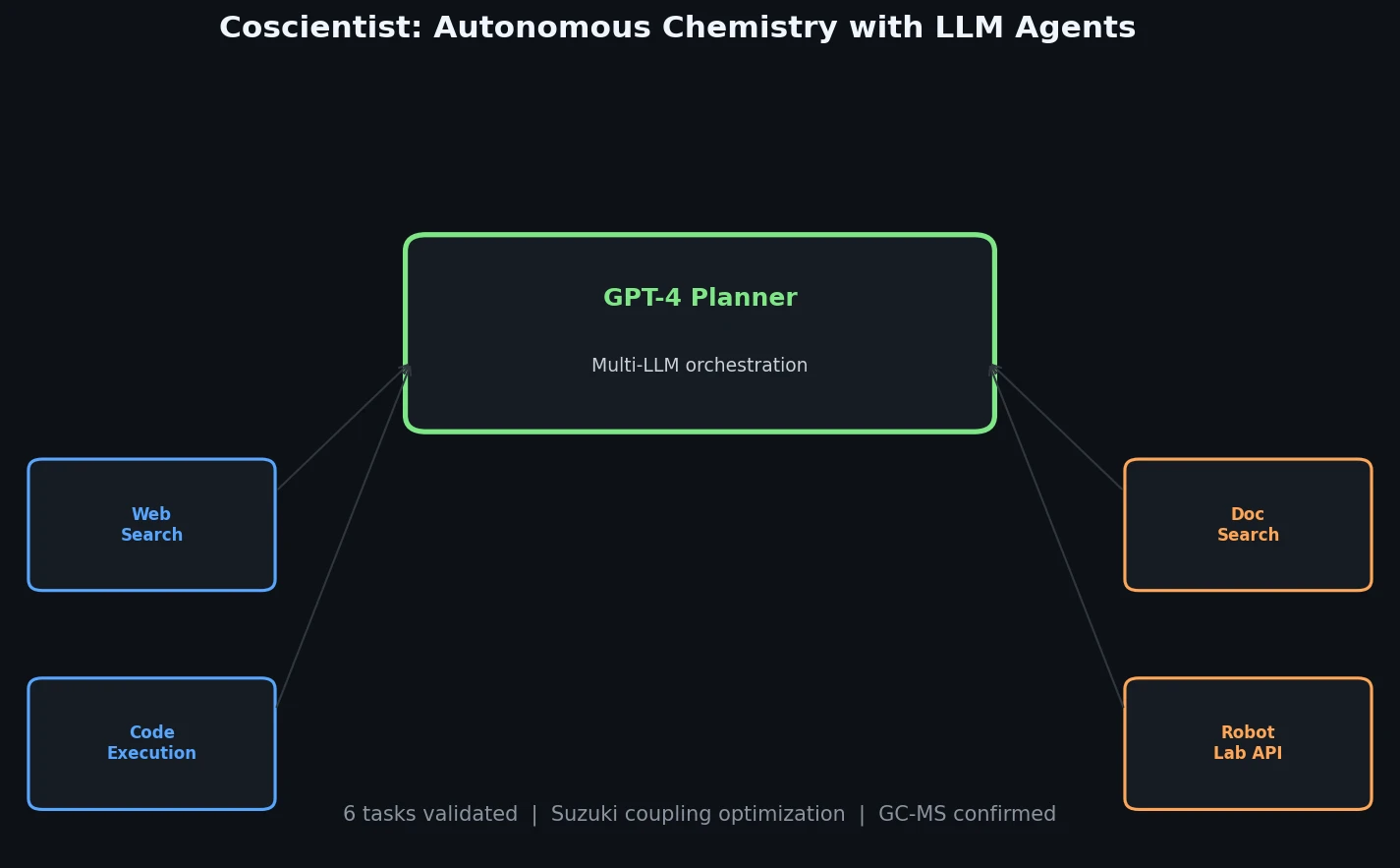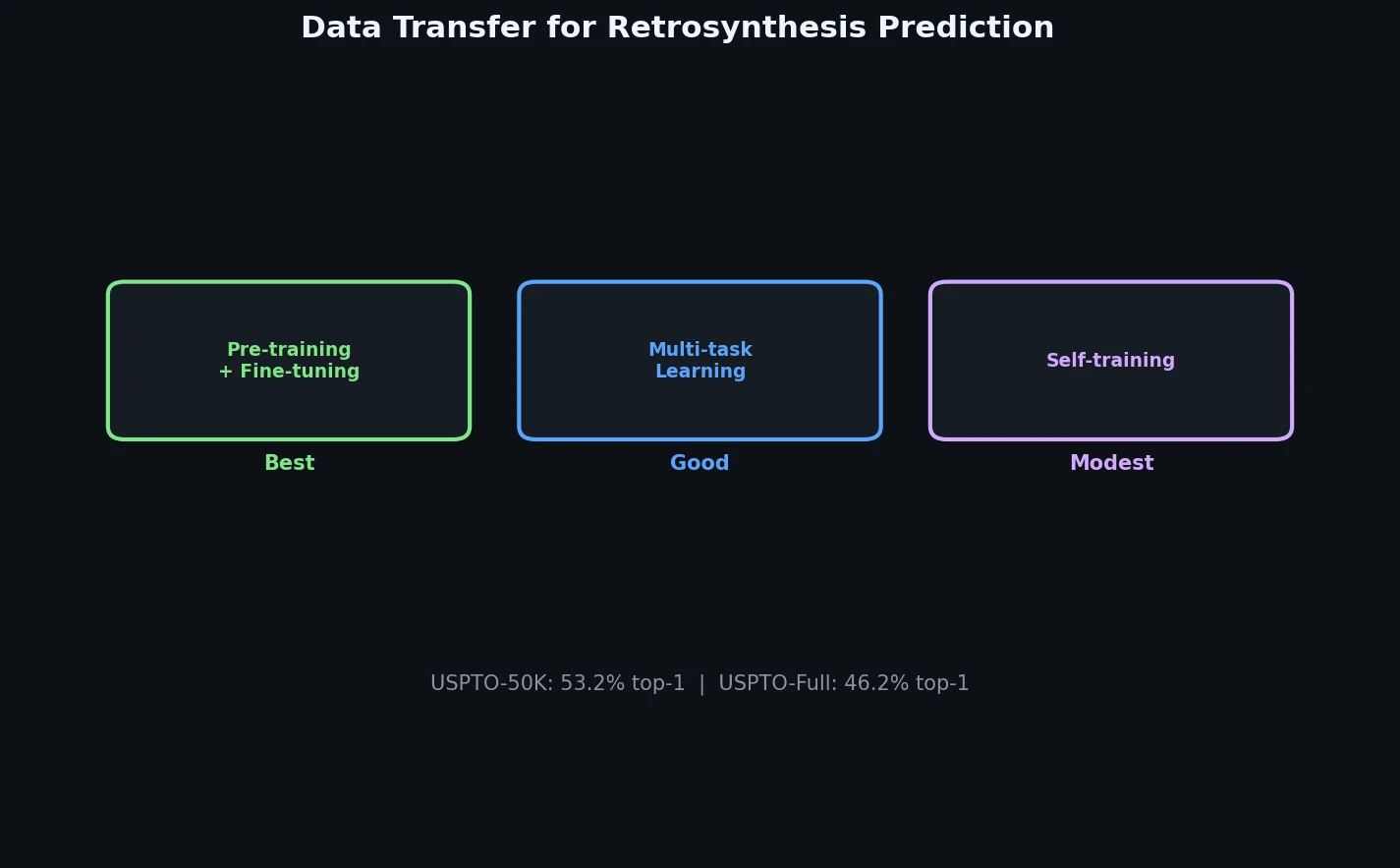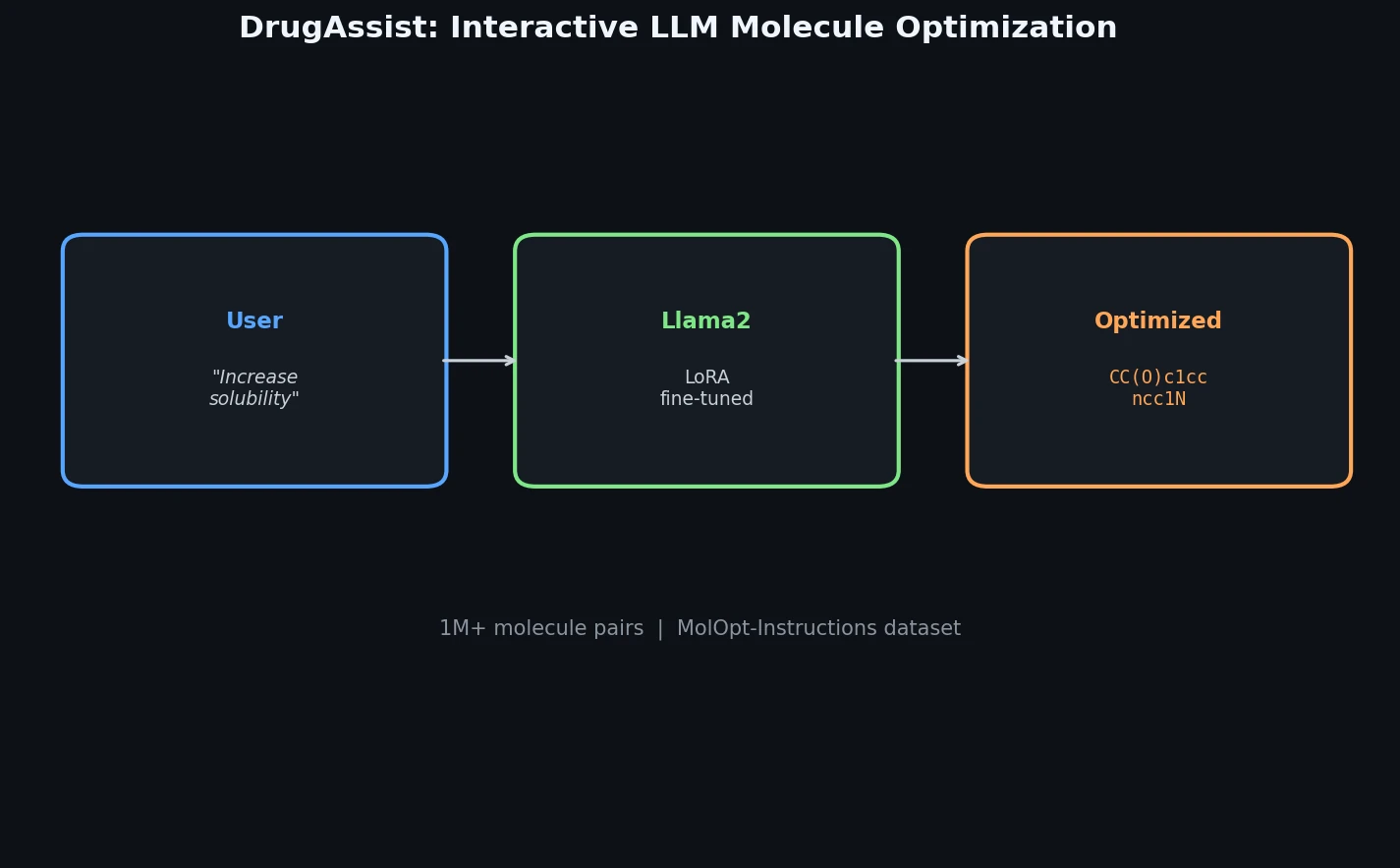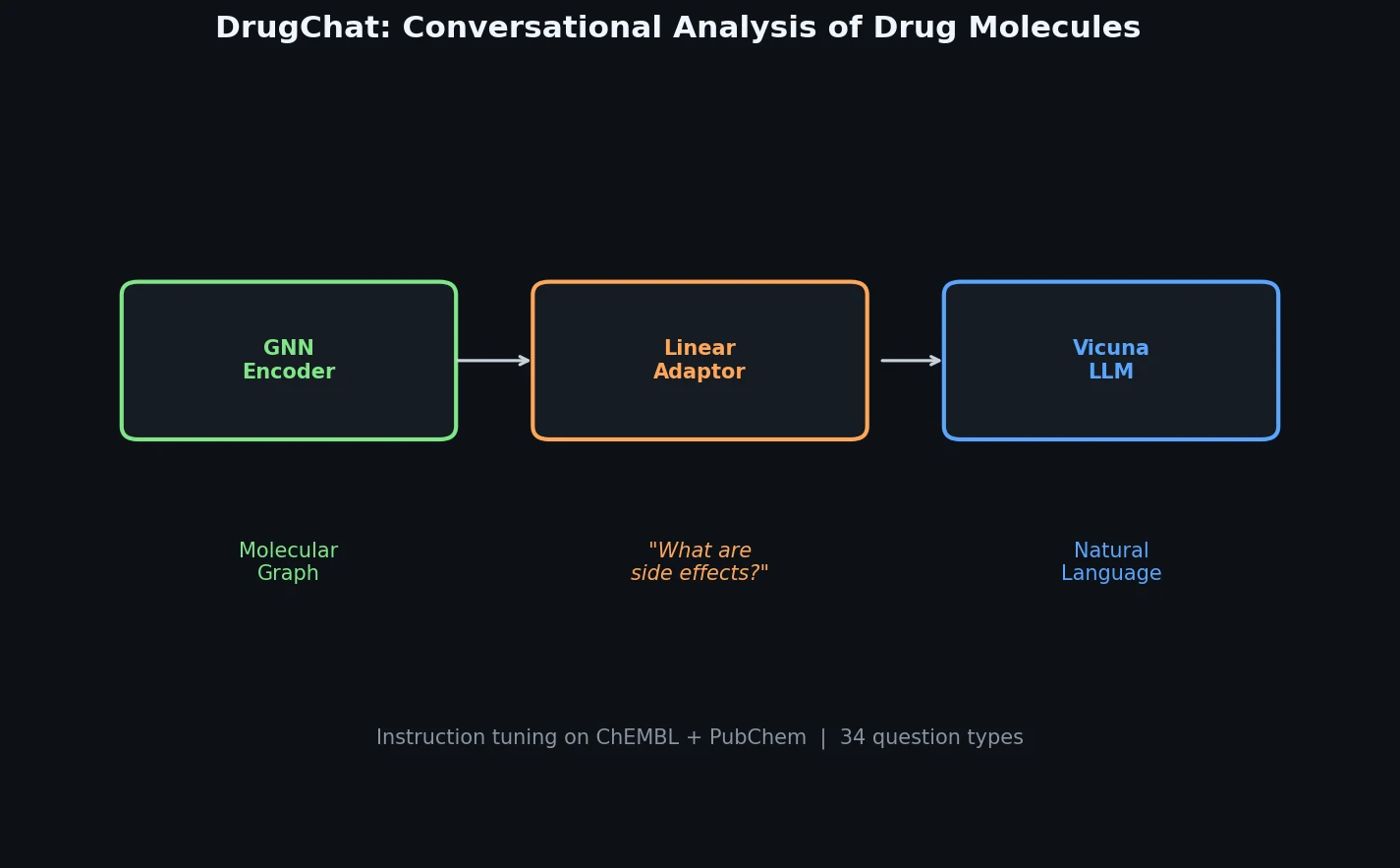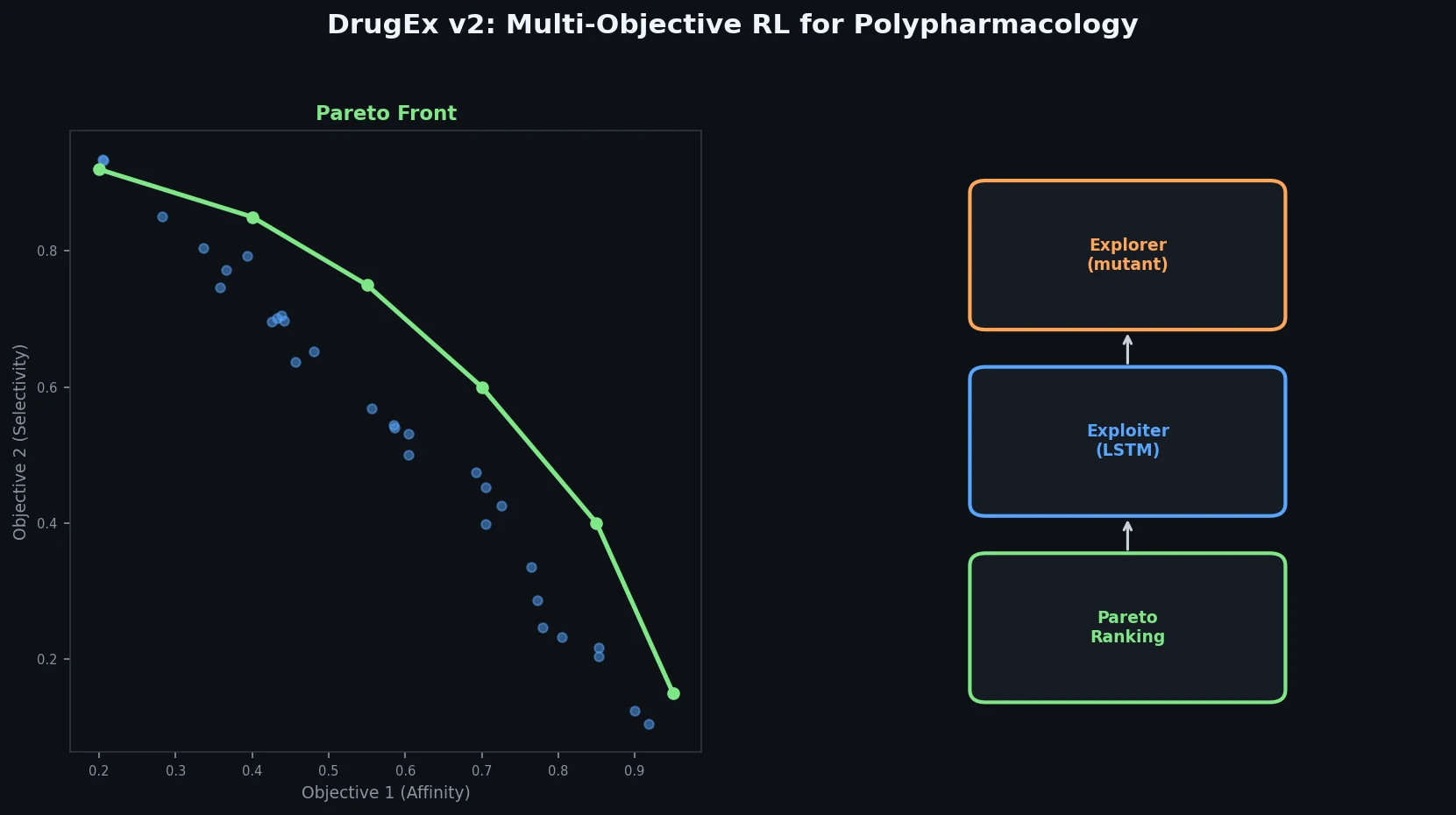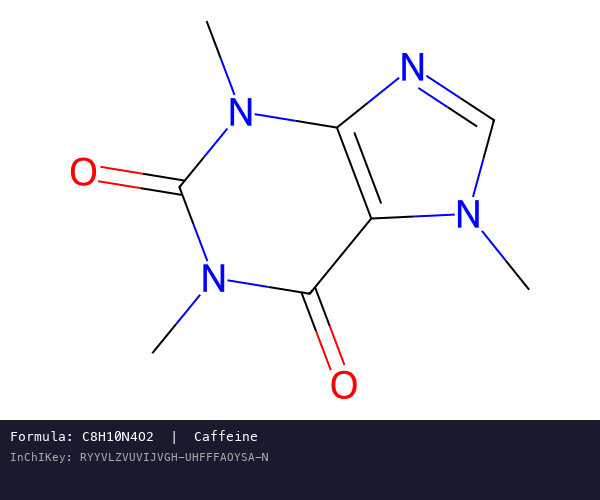
InChI: The International Chemical Identifier
InChI (International Chemical Identifier) is an open standard from IUPAC that represents molecular structures as hierarchical, layered strings optimized for database interoperability, unique identification, and web search via its hashed InChIKey.