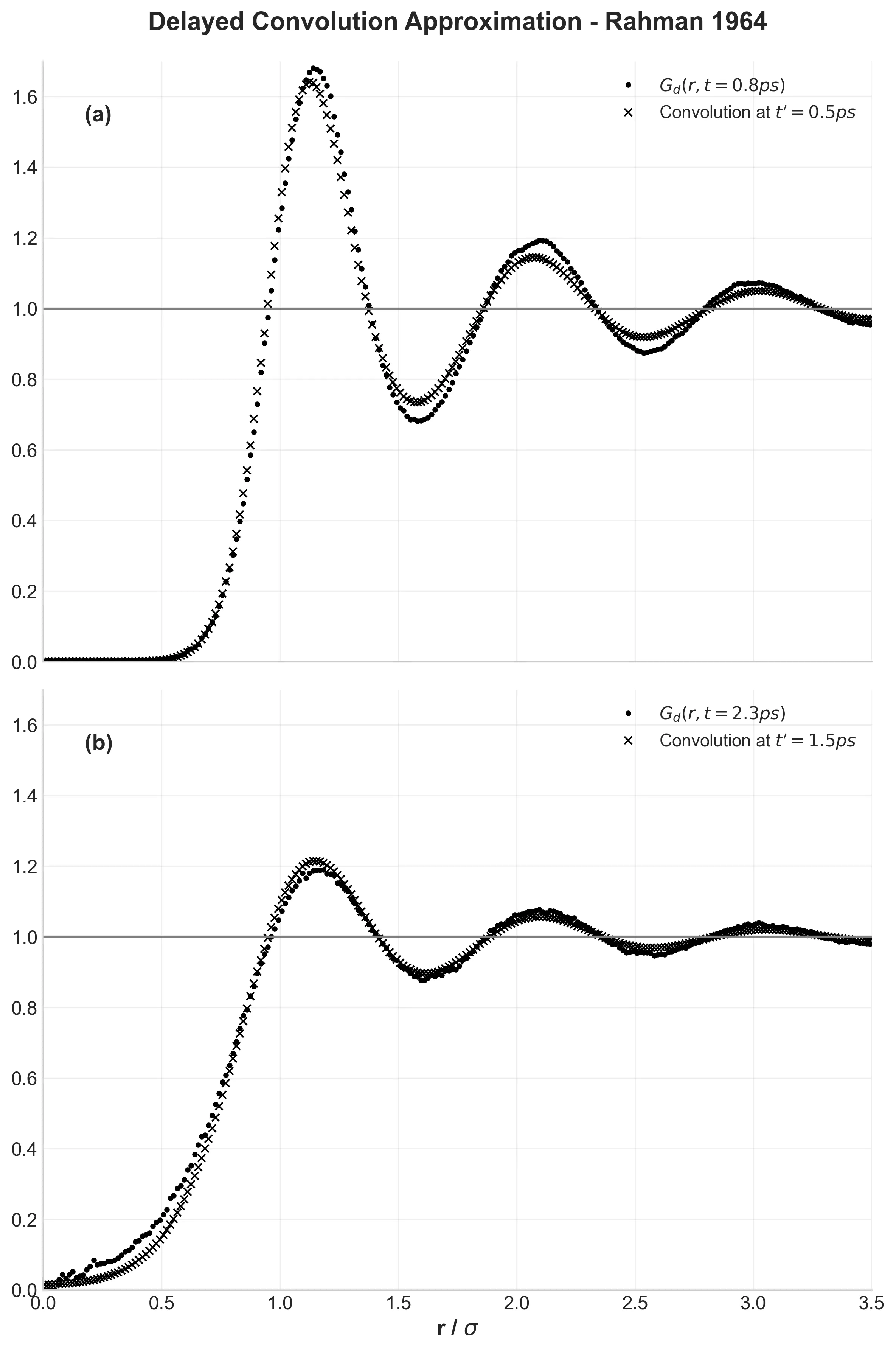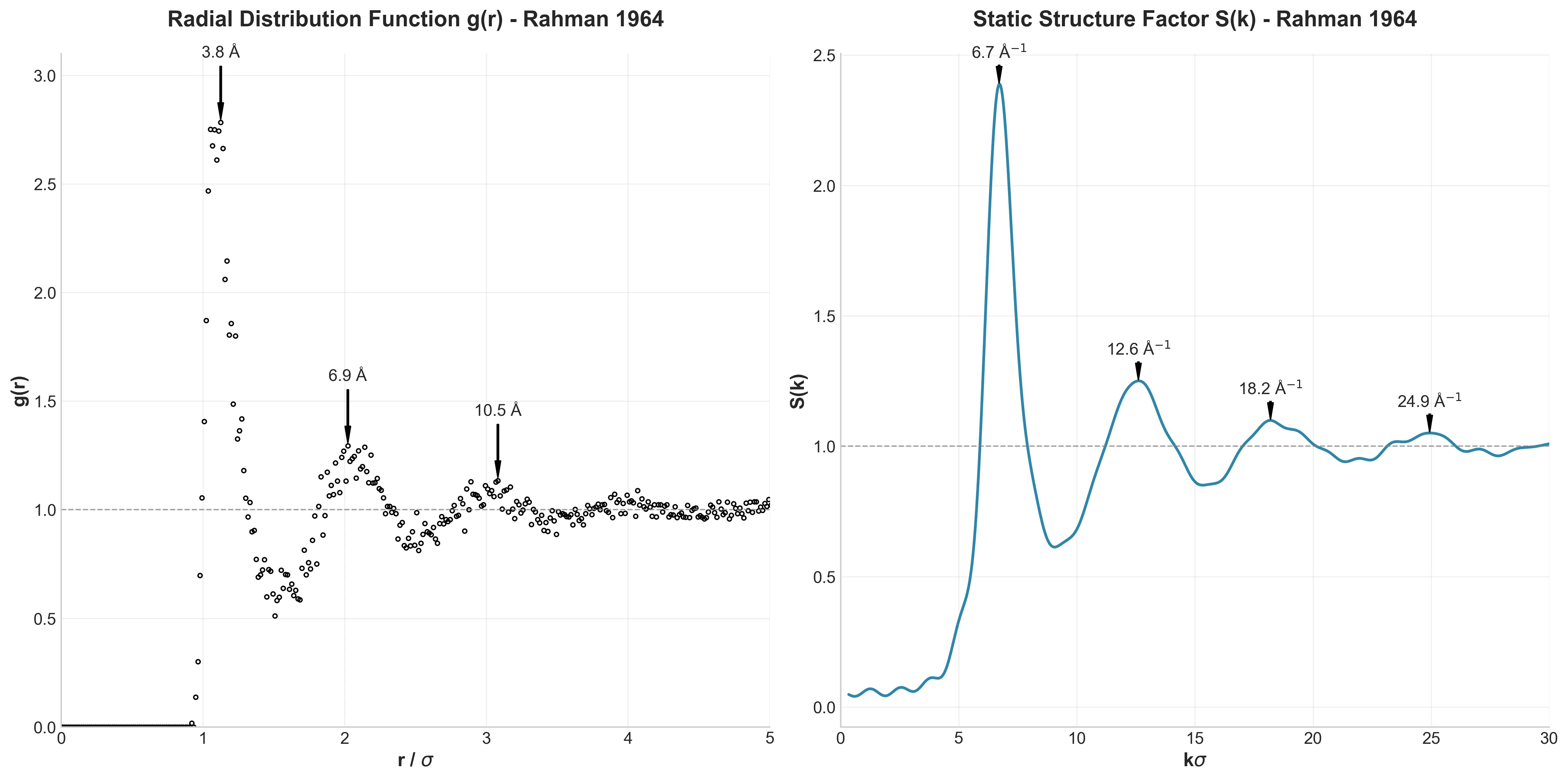
MOFFlow: Flow Matching for MOF Structure Prediction
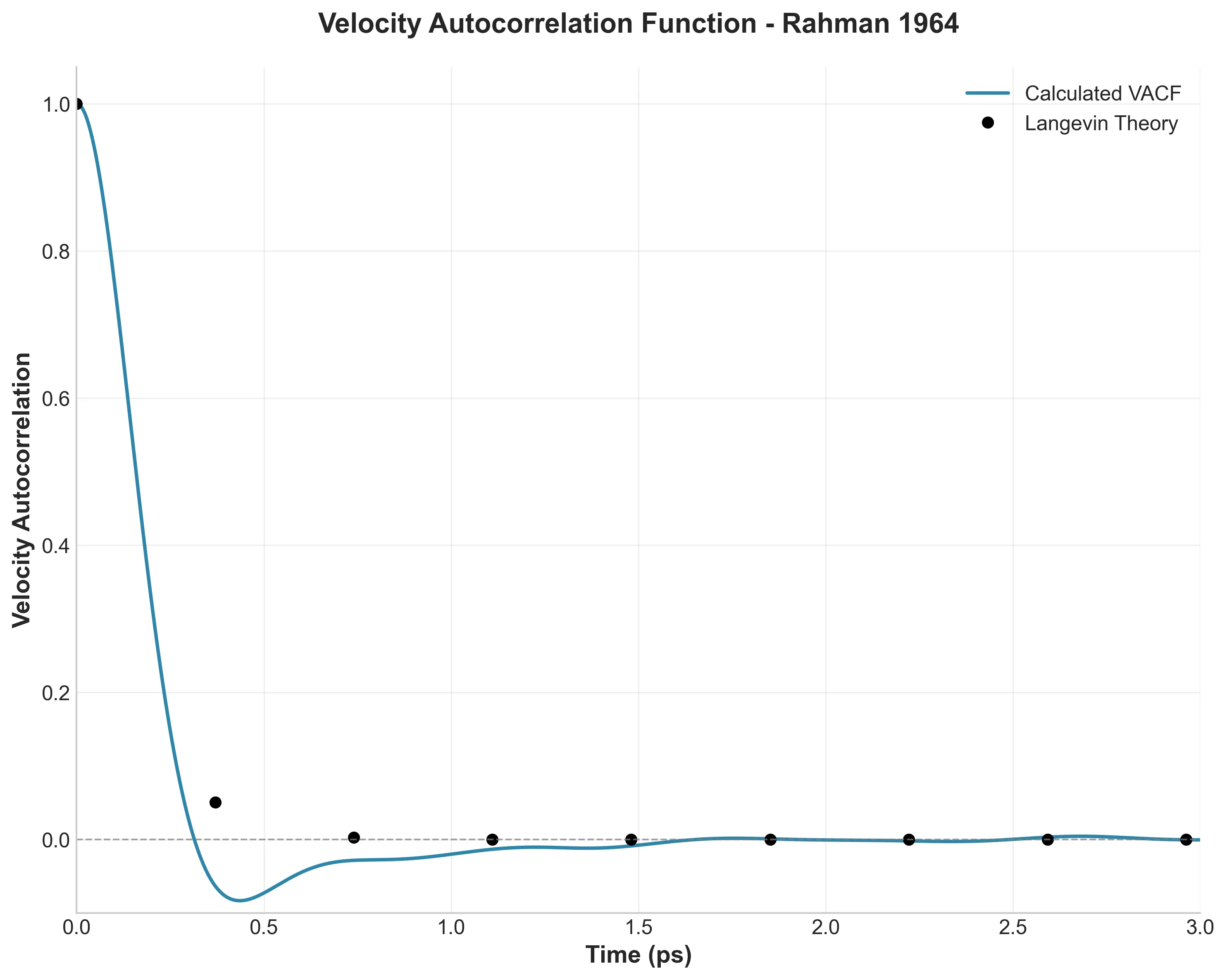
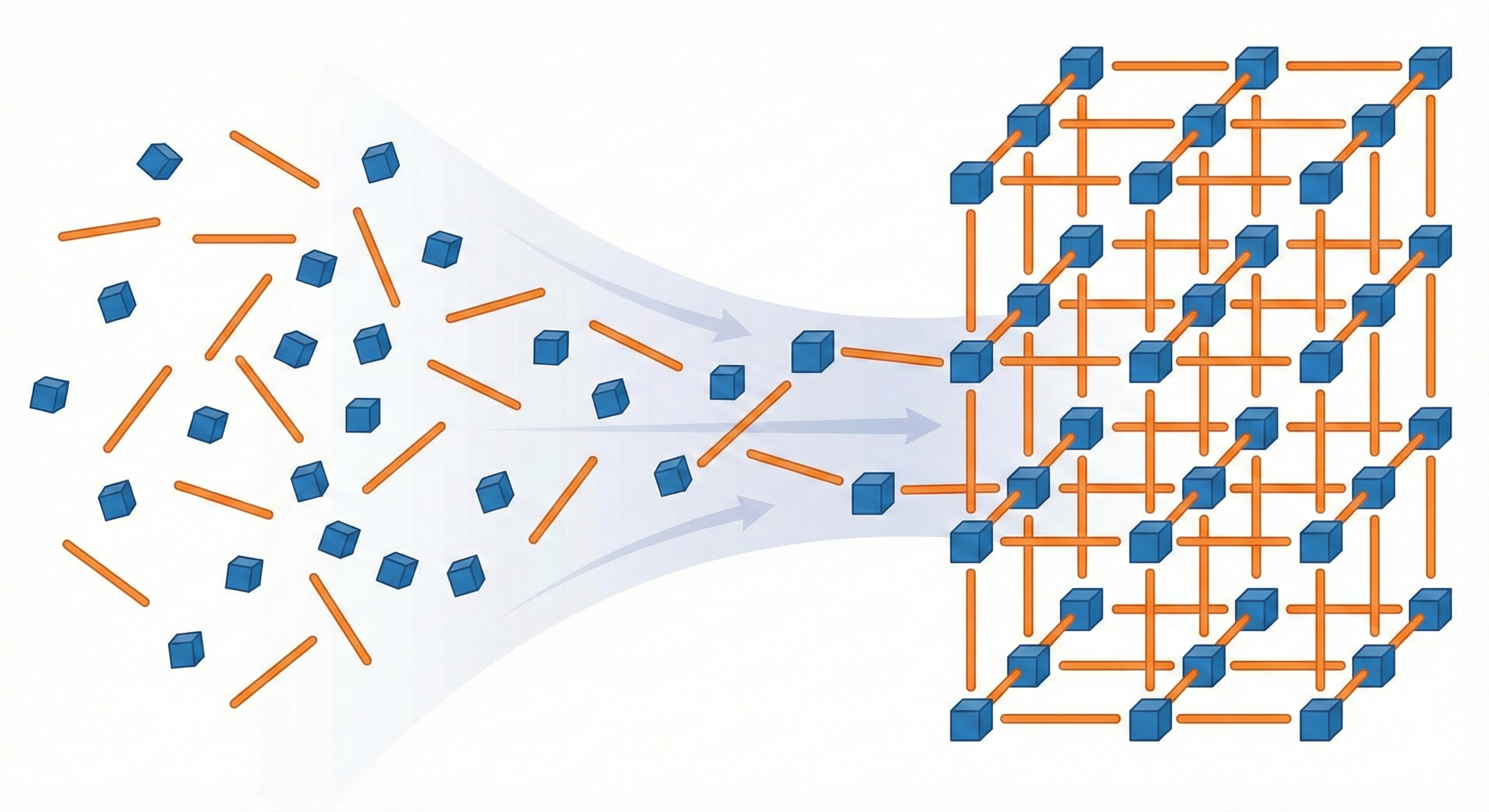
MOFFlow is the first deep generative model tailored for Metal-Organic Framework (MOF) structure prediction. It utilizes Riemannian flow matching on SE(3) to assemble rigid building blocks (metal nodes and organic linkers), achieving significantly higher accuracy and scalability than atom-based methods on large systems.