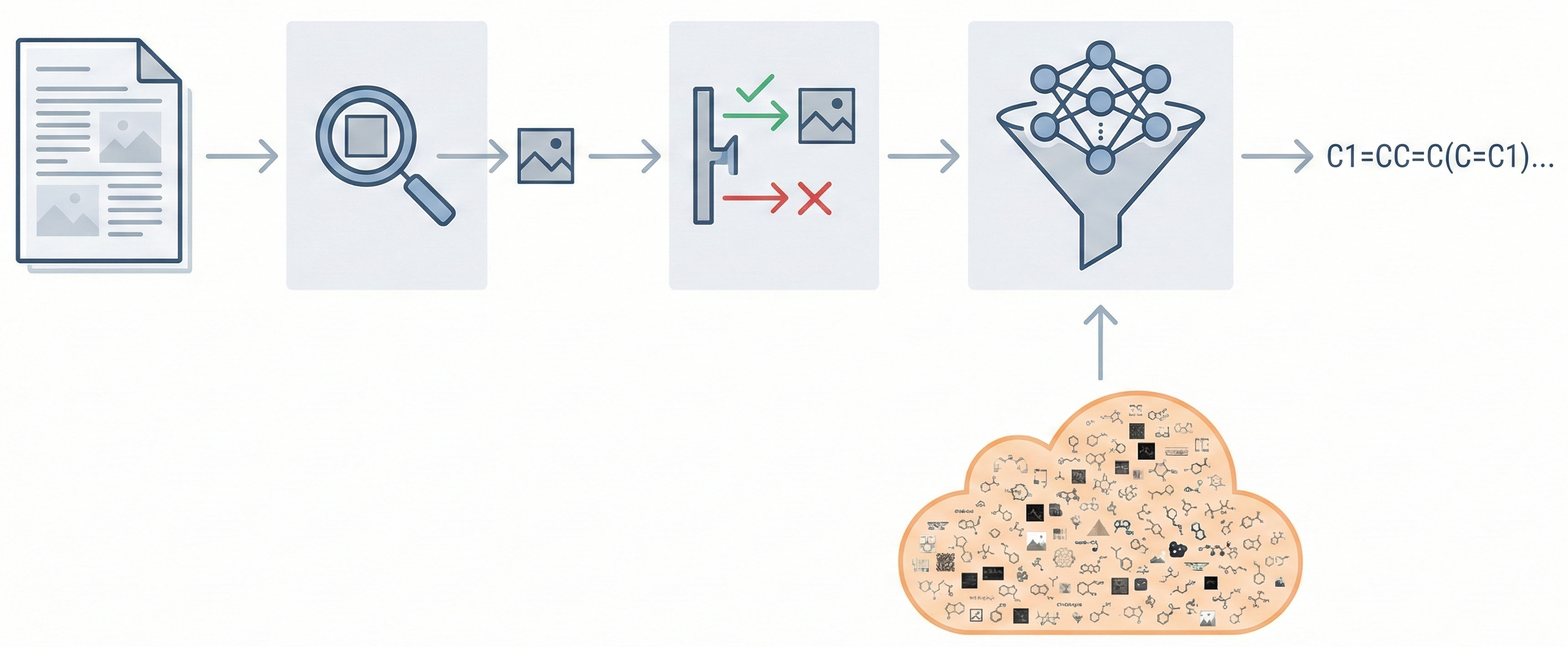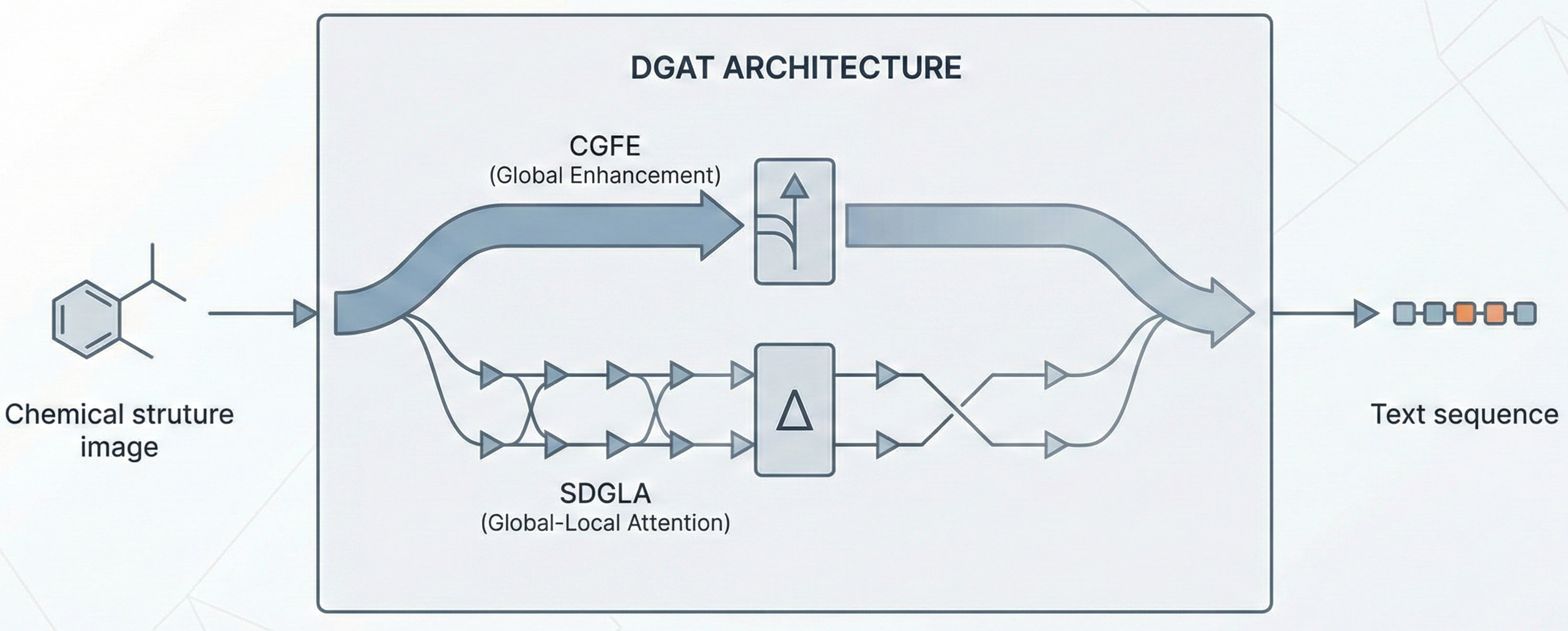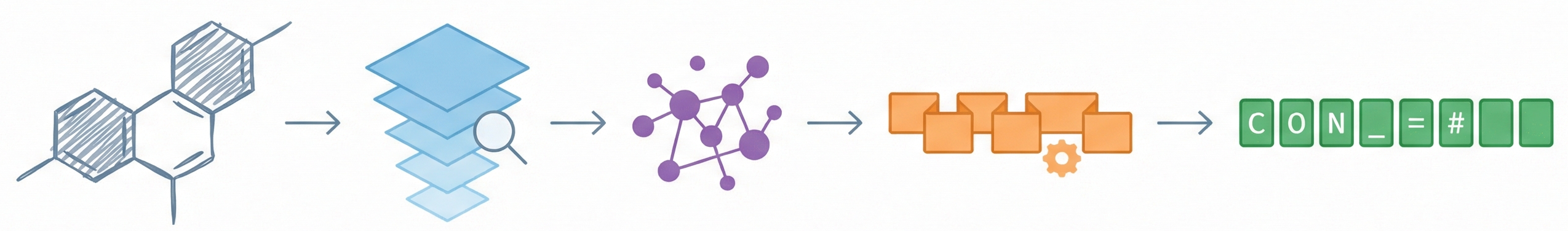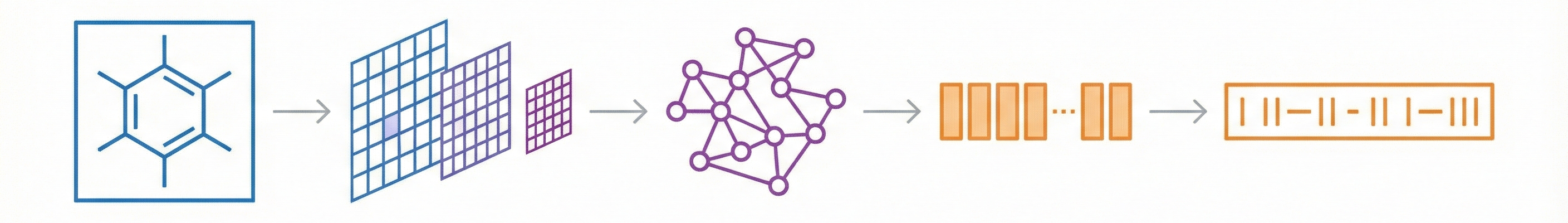
Molecular Sets (MOSES): A Generative Modeling Benchmark
MOSES introduces a comprehensive benchmarking platform for molecular generative models, offering standardized datasets, evaluation metrics, and baselines. By providing a unified measuring stick, it aims to resolve reproducibility challenges in chemical distribution learning.