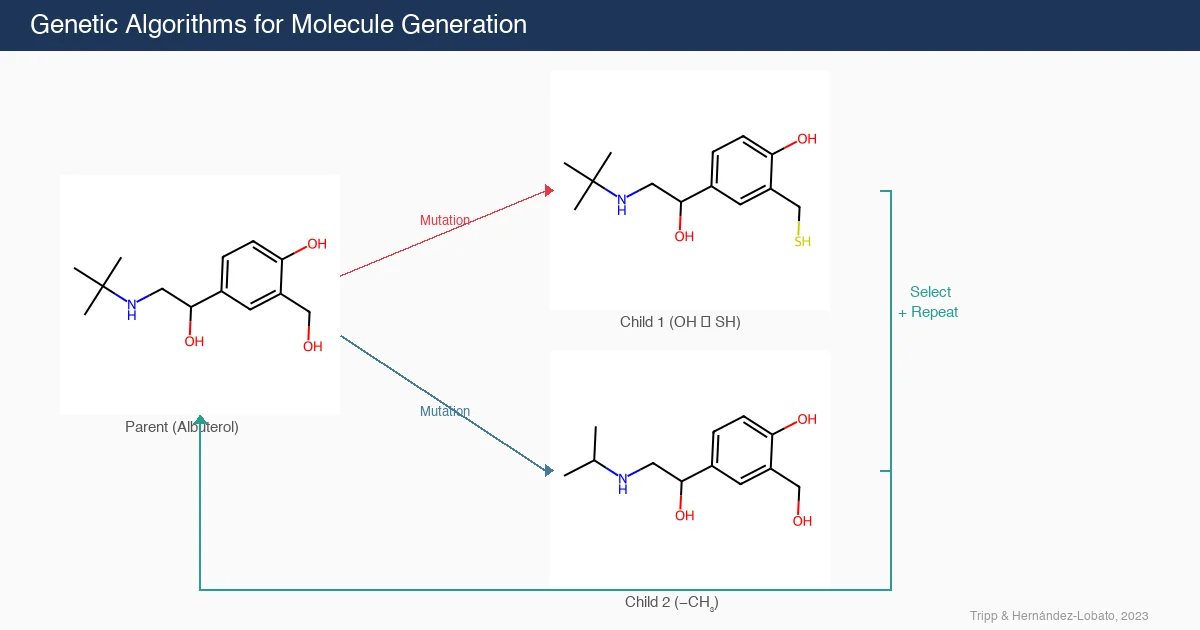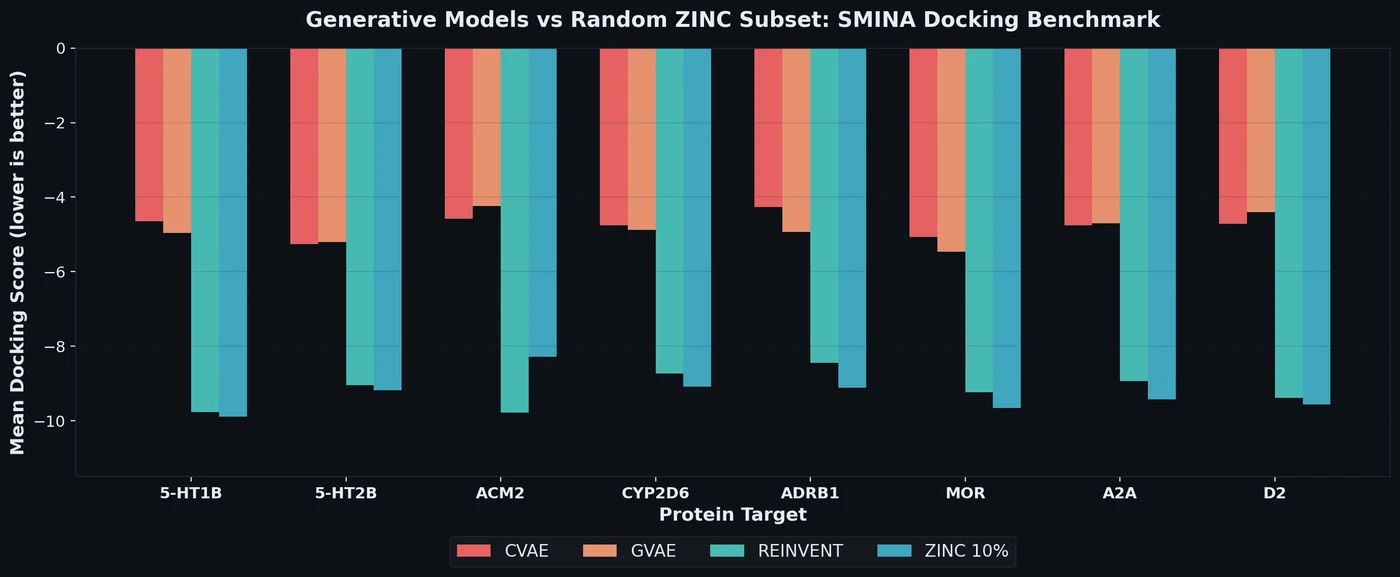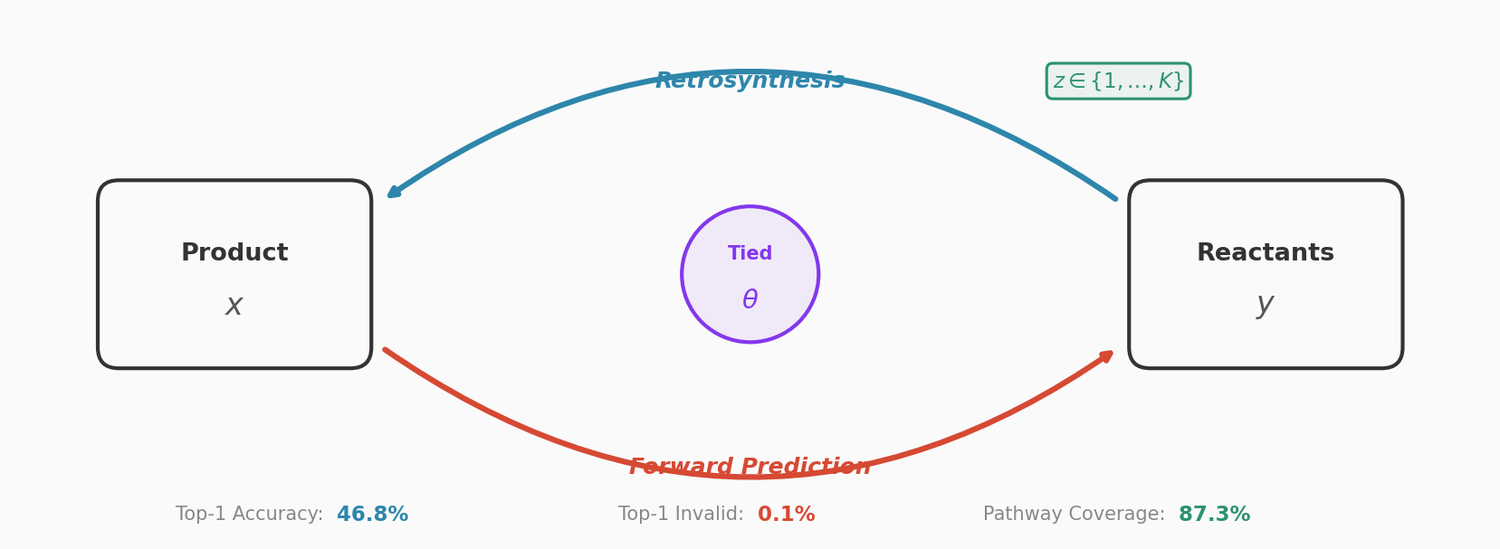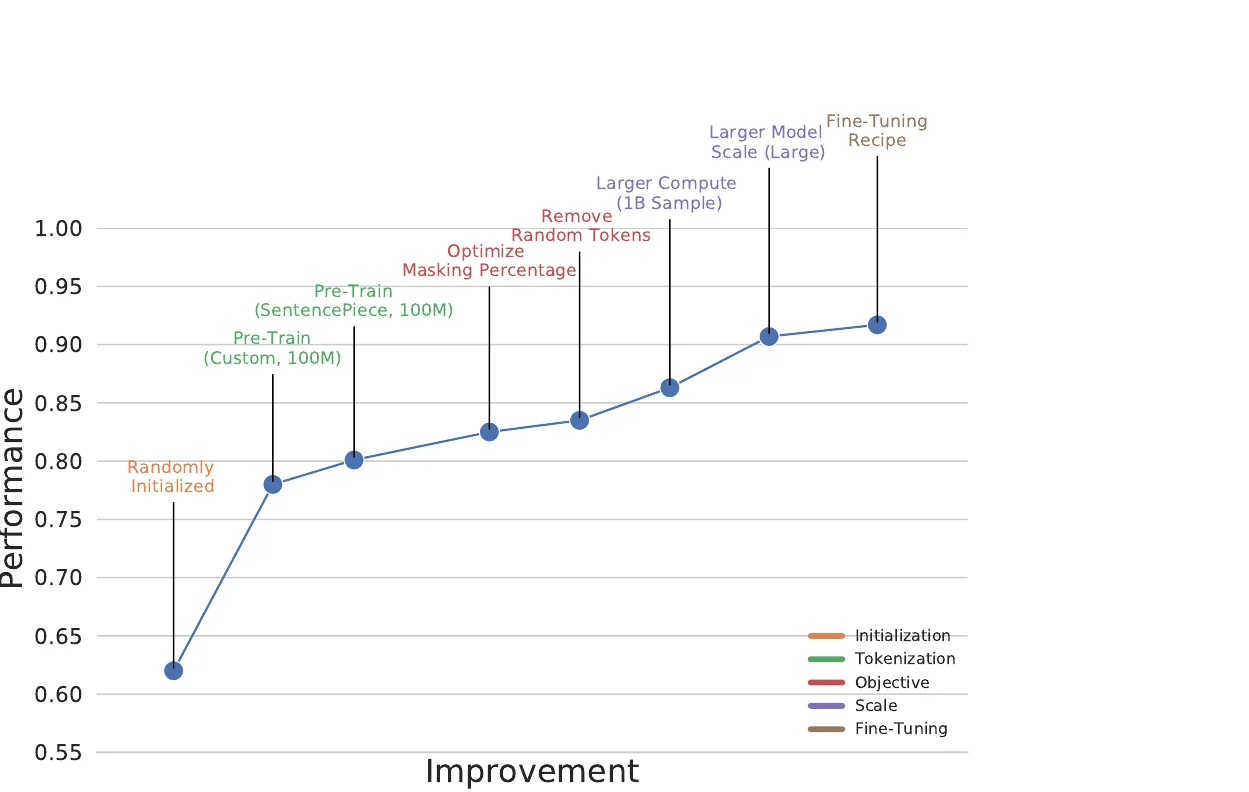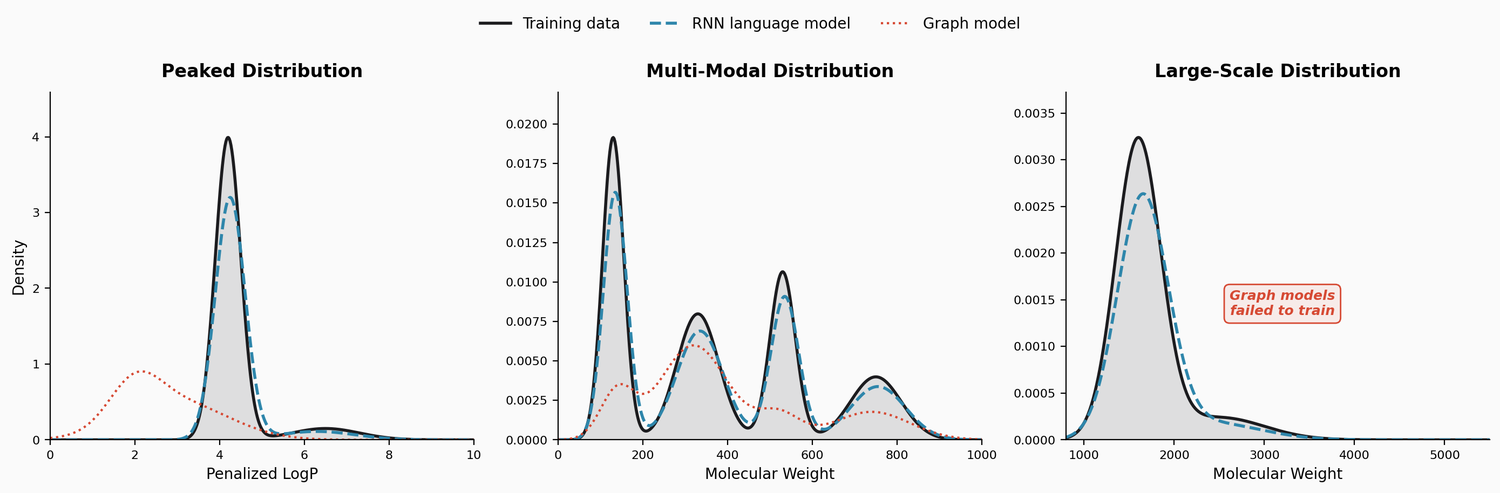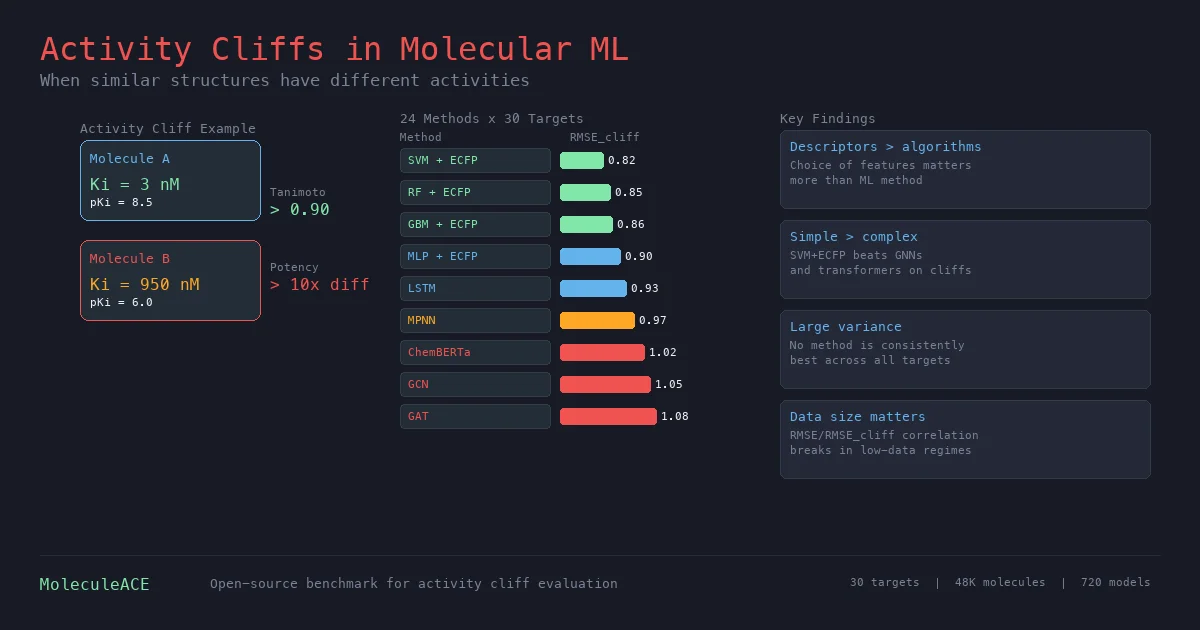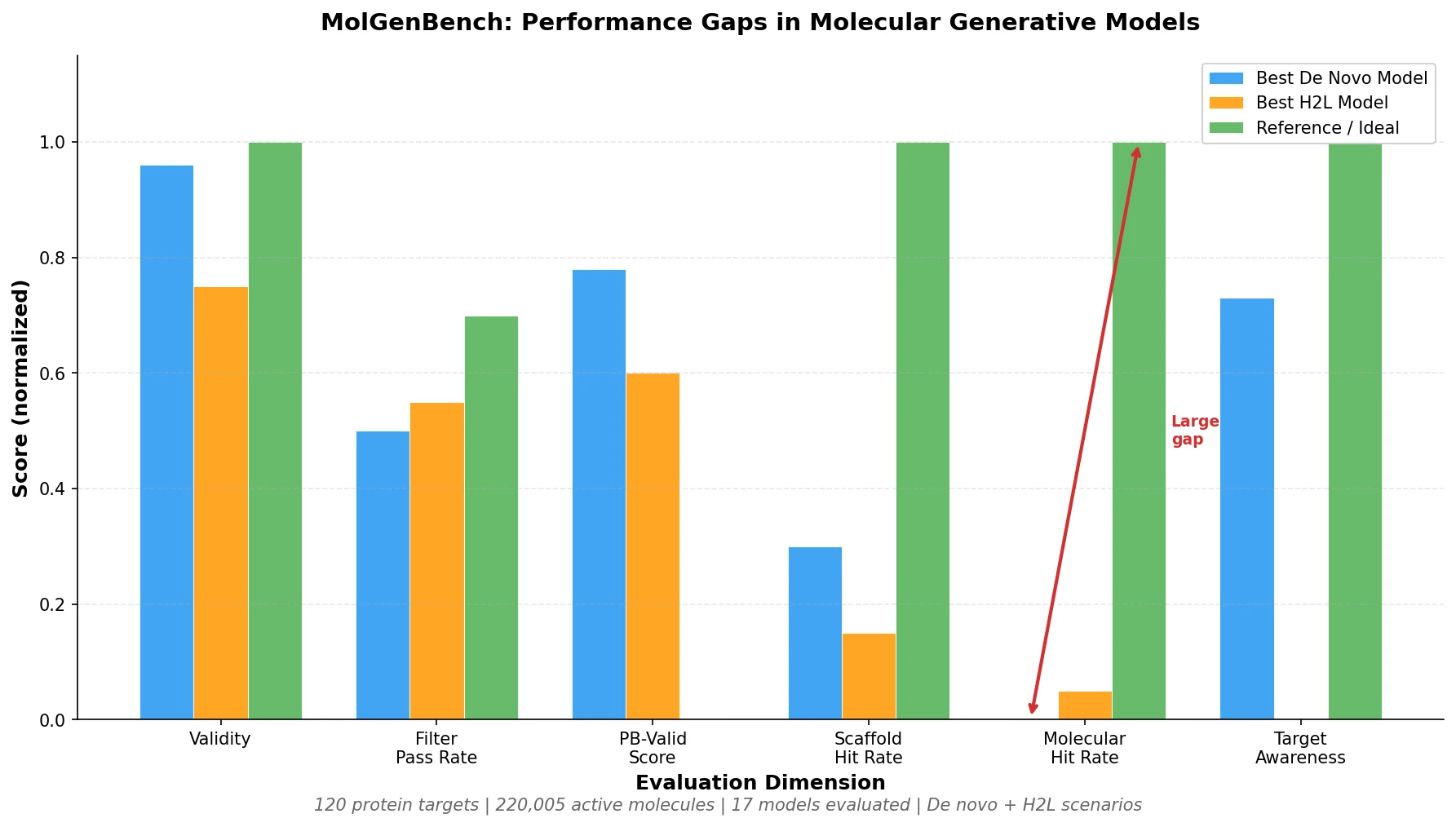
MolGenBench: Benchmarking Molecular Generative Models
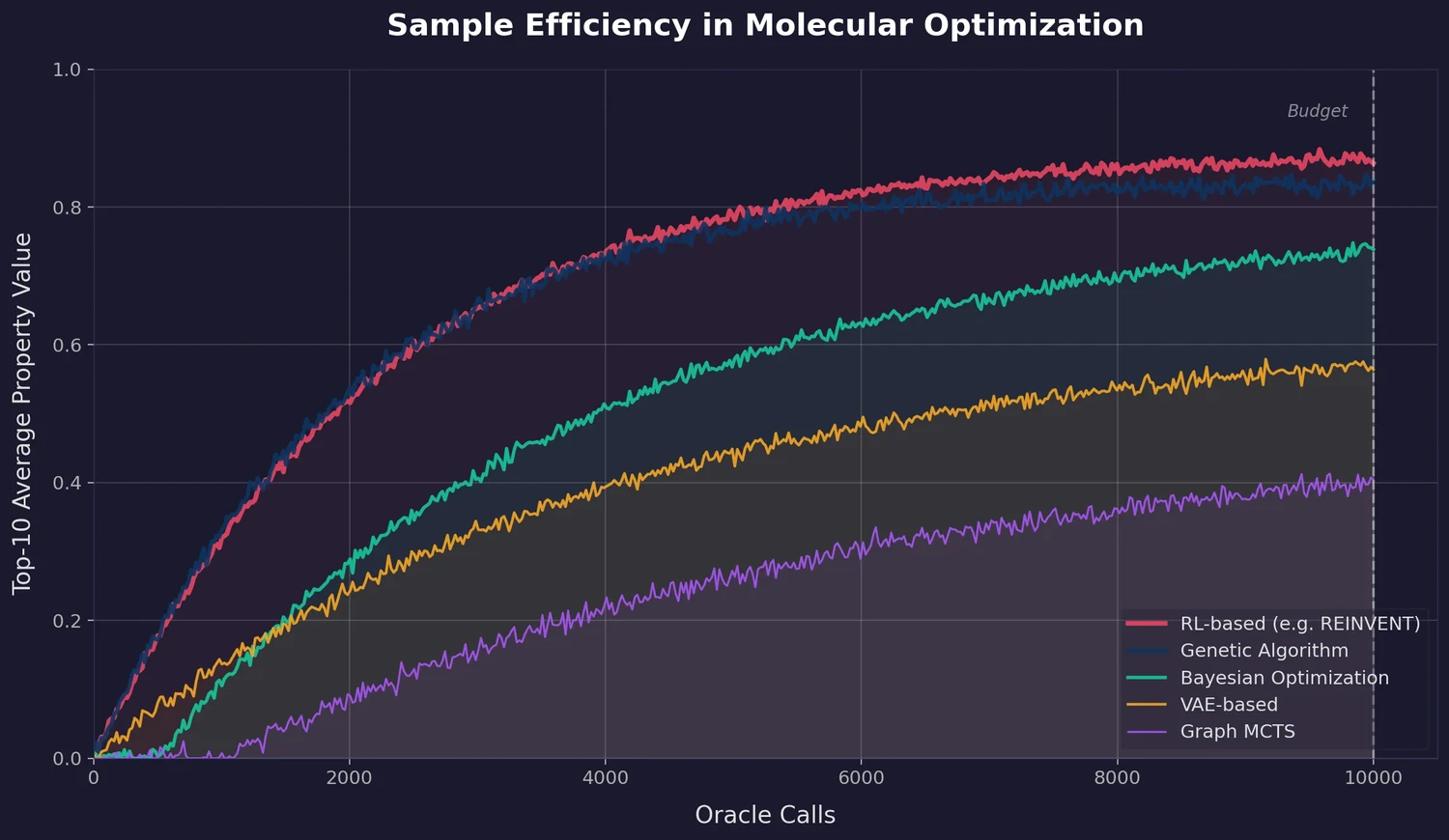
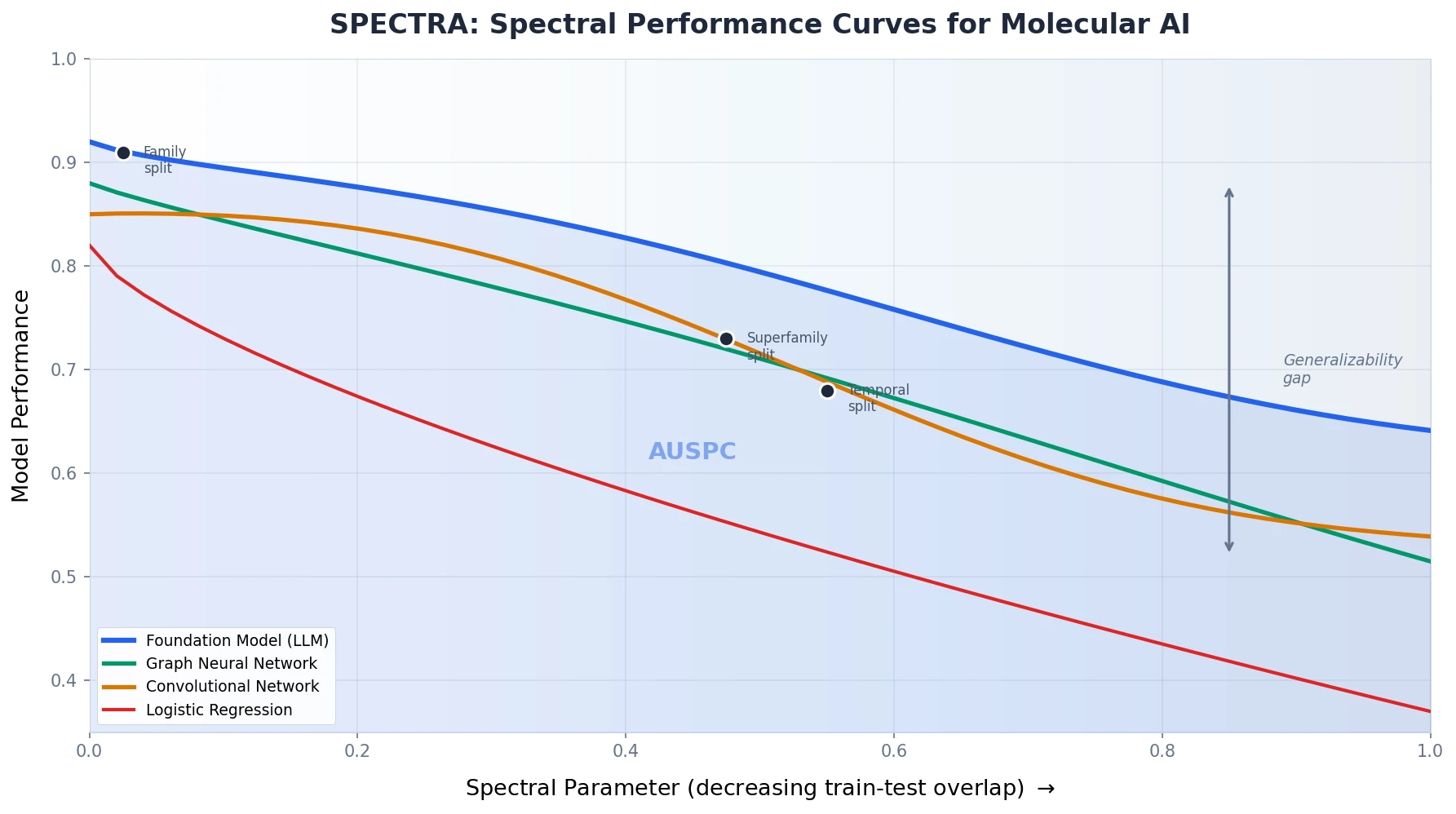
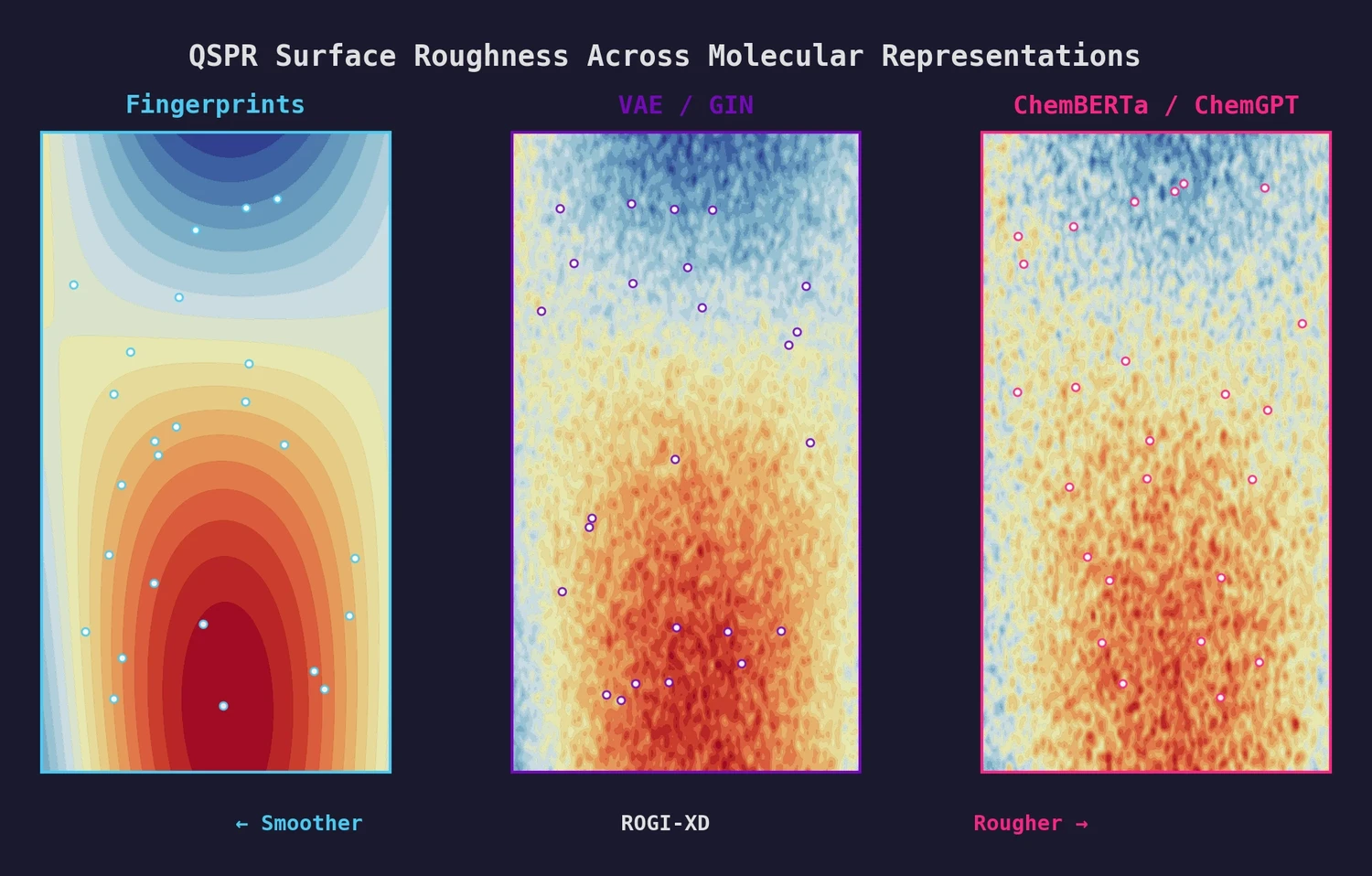
MolGenBench introduces a comprehensive benchmark for evaluating molecular generative models in realistic drug discovery settings, spanning de novo design and hit-to-lead optimization across 120 protein targets with 220,005 experimentally validated actives.