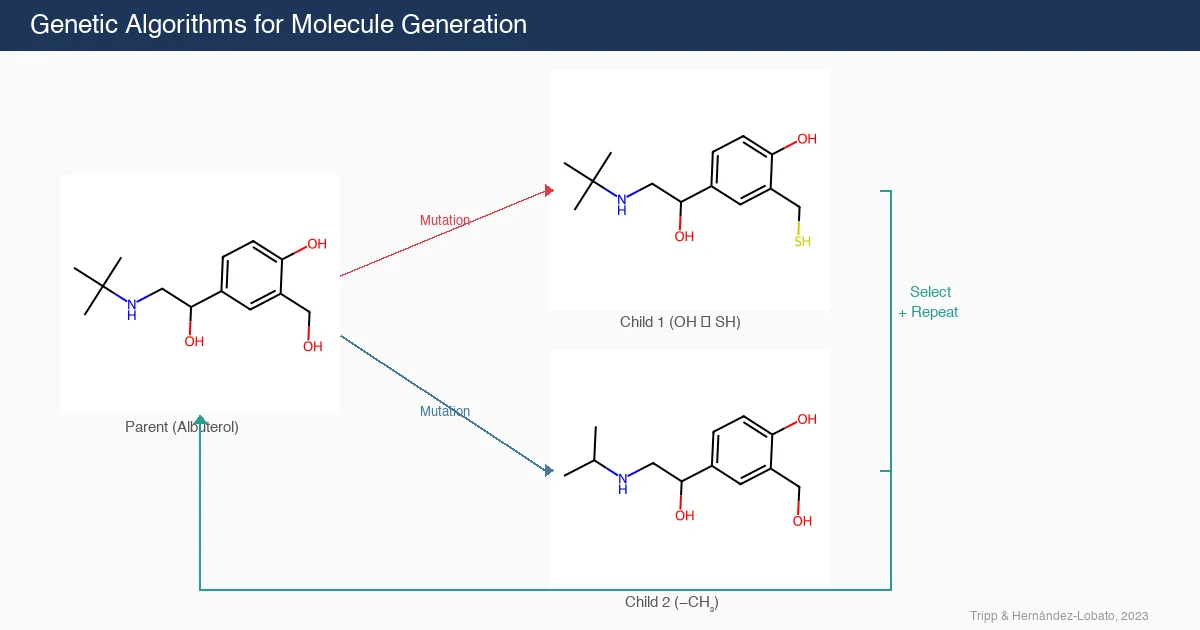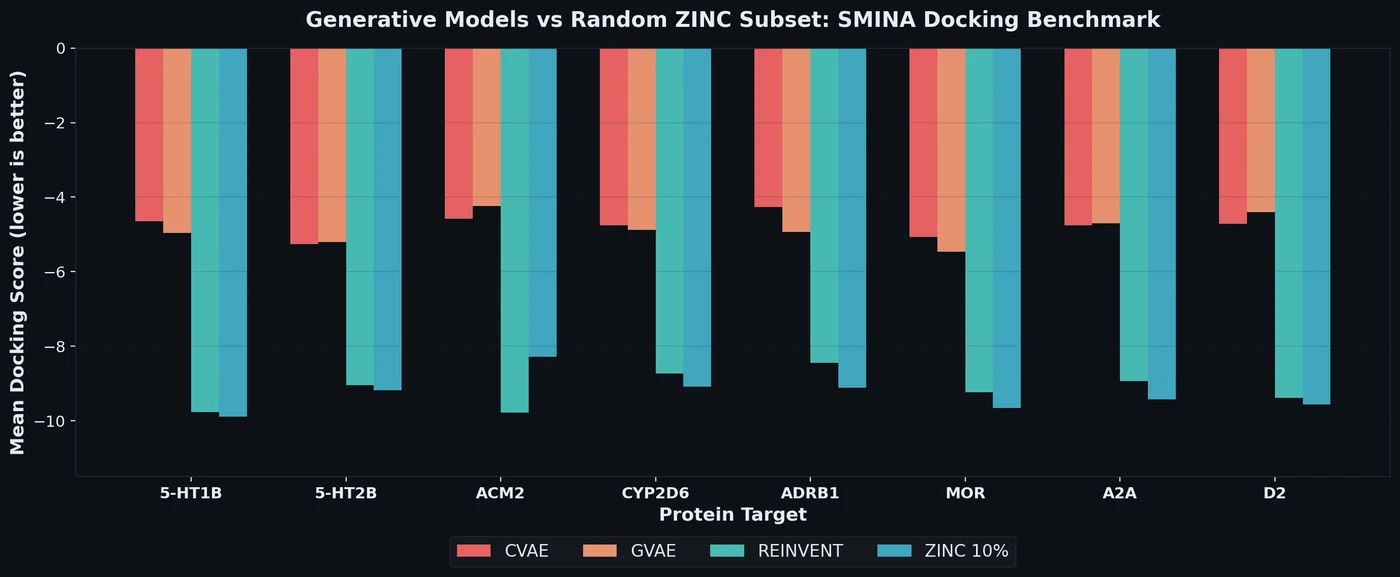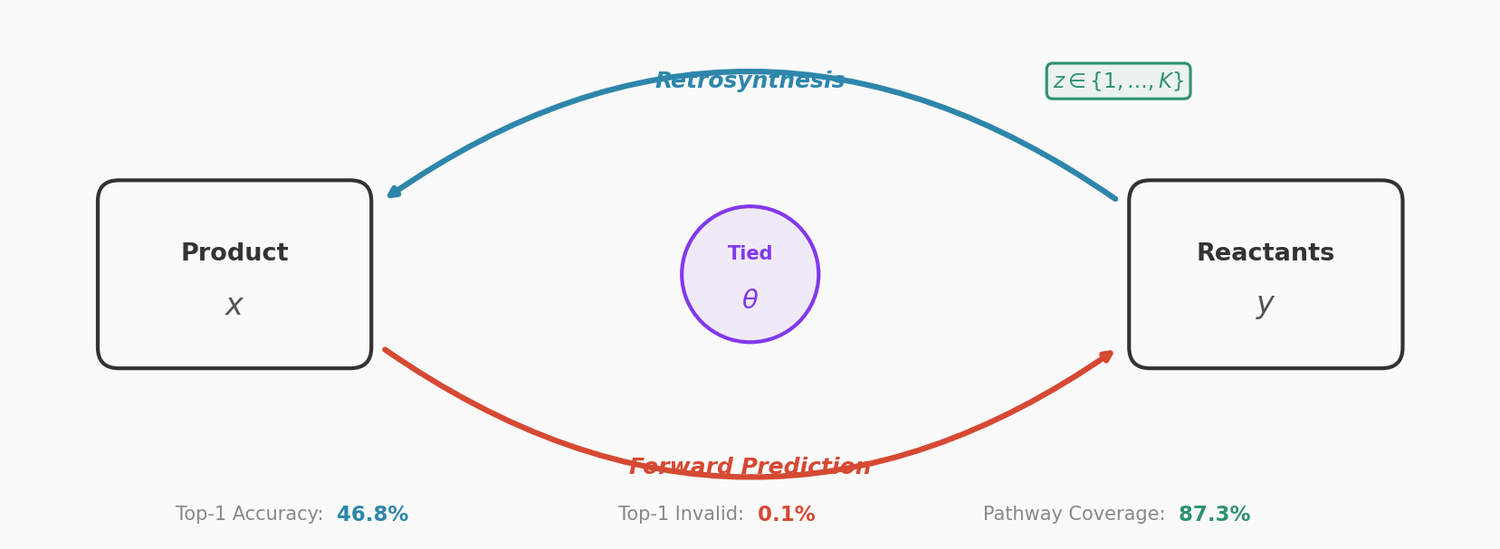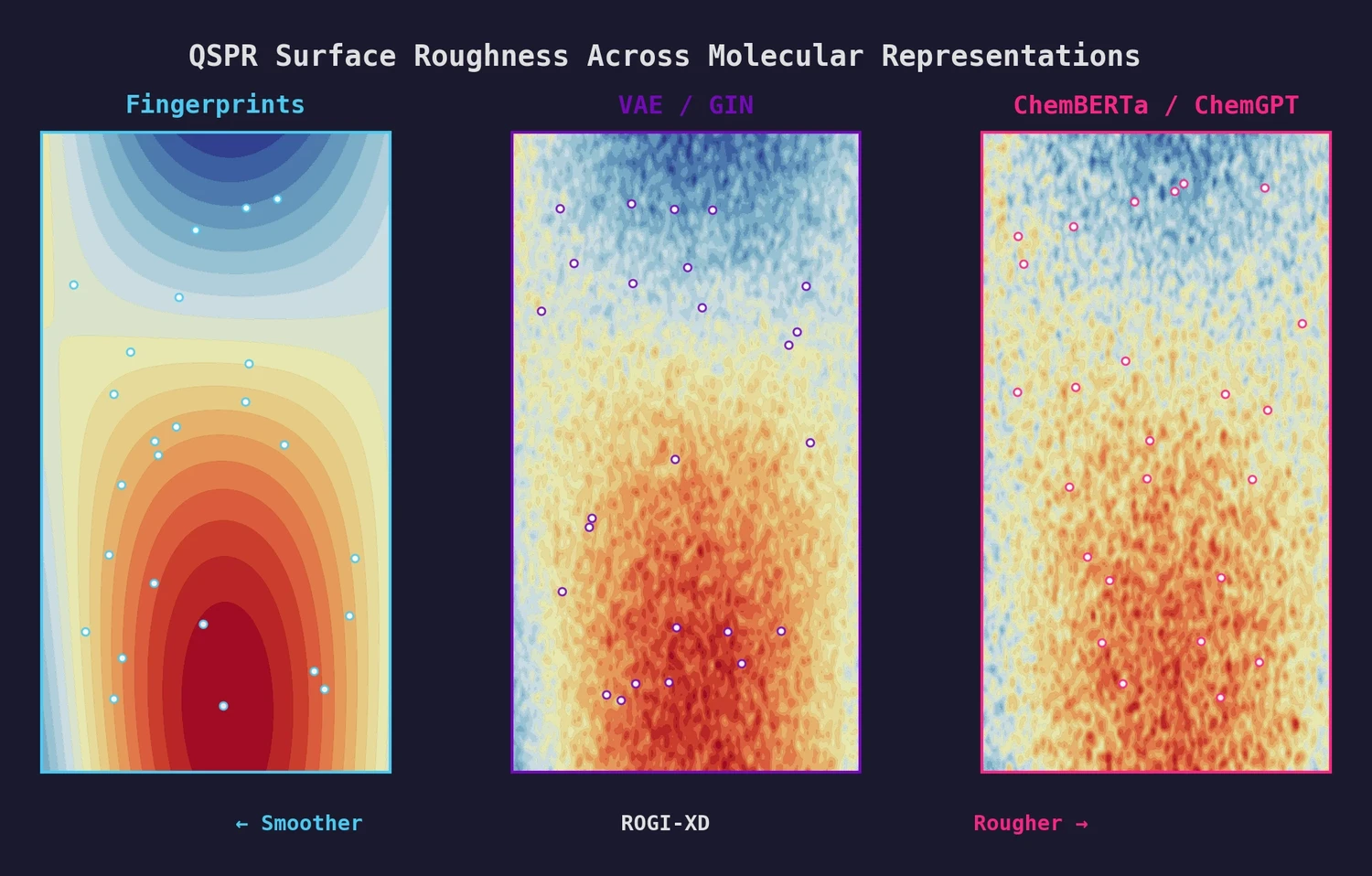
ROGI-XD: Roughness of Pretrained Molecular Representations
This paper introduces ROGI-XD, a reformulation of the ROuGhness Index that enables fair comparison of QSPR surface roughness across molecular representations of different dimensionalities. Evaluating VAE, GIN, ChemBERTa, and ChemGPT representations, the authors show that pretrained chemical models do not produce smoother structure-property landscapes than simple molecular fingerprints or descriptors.