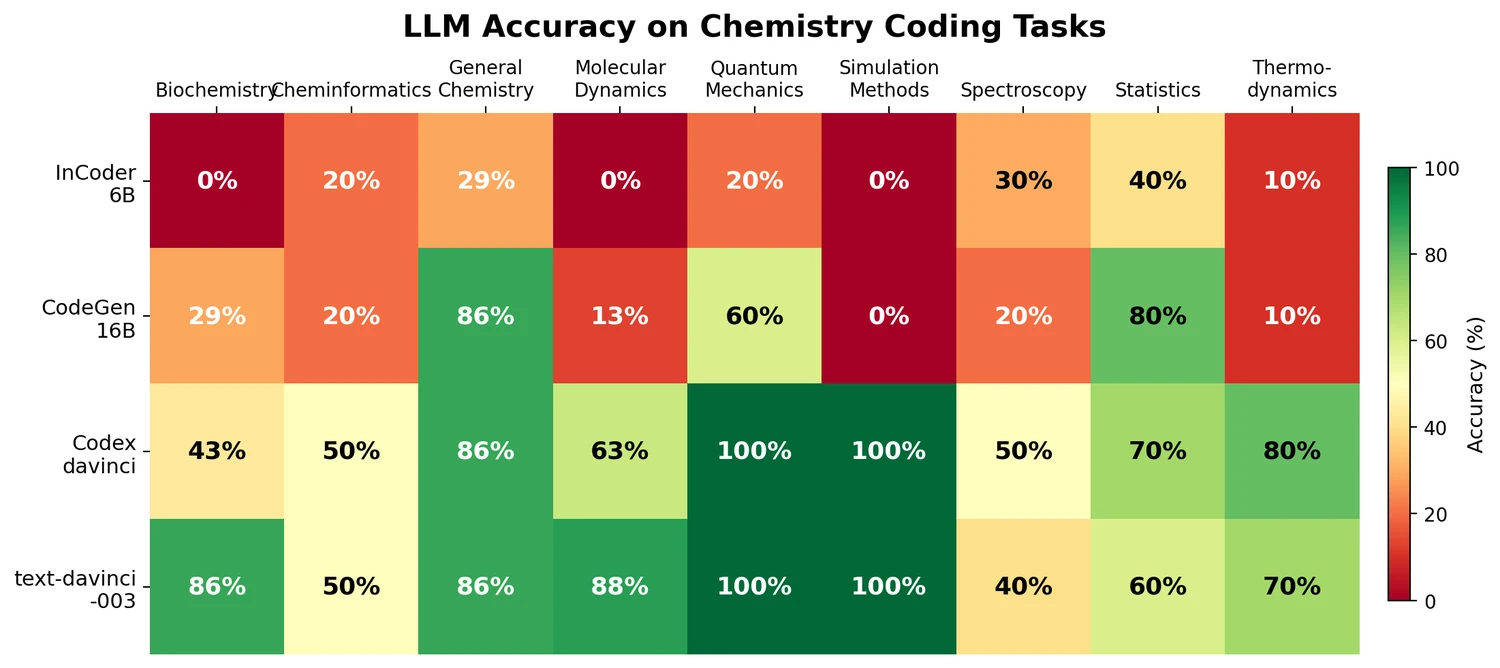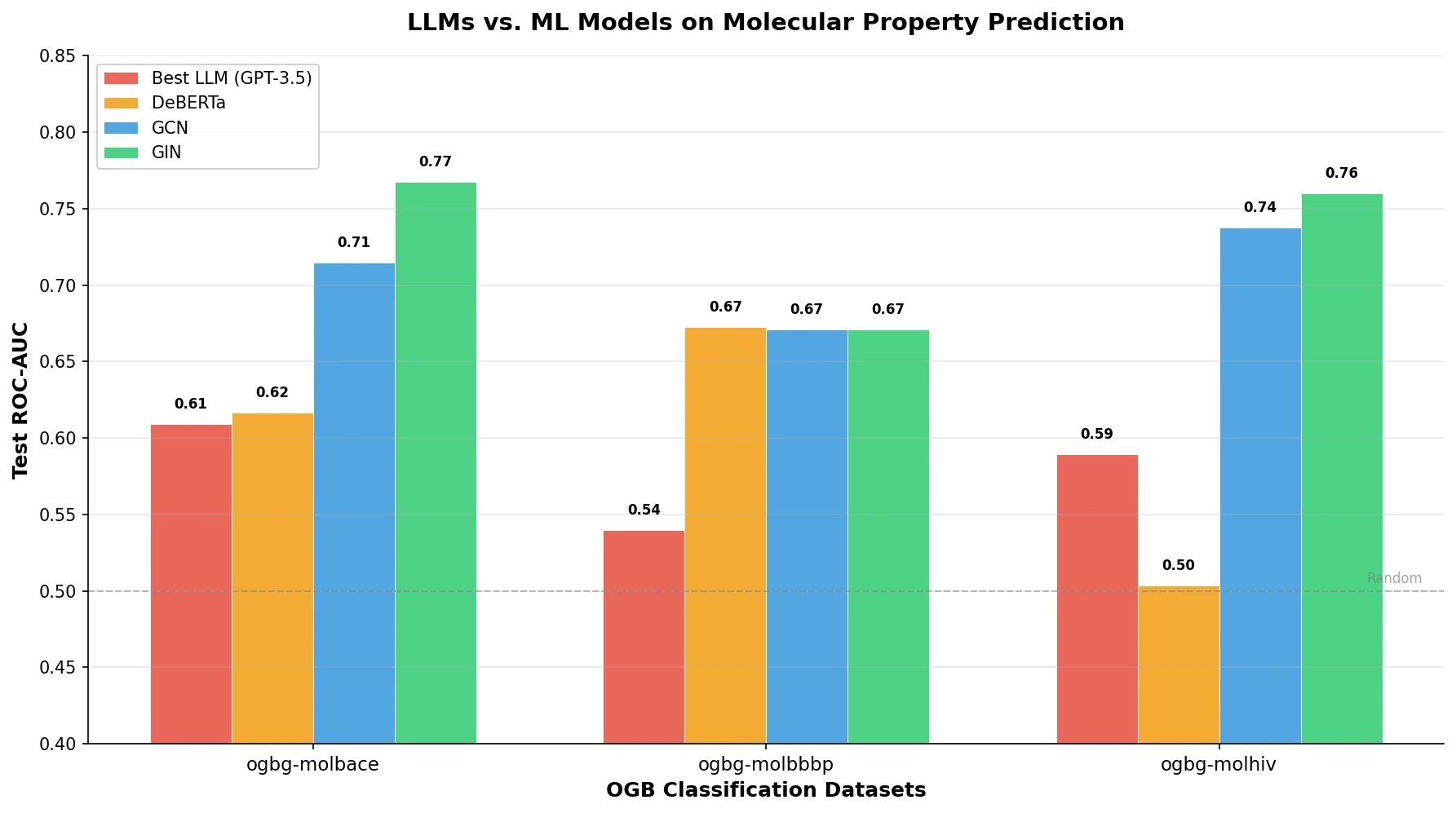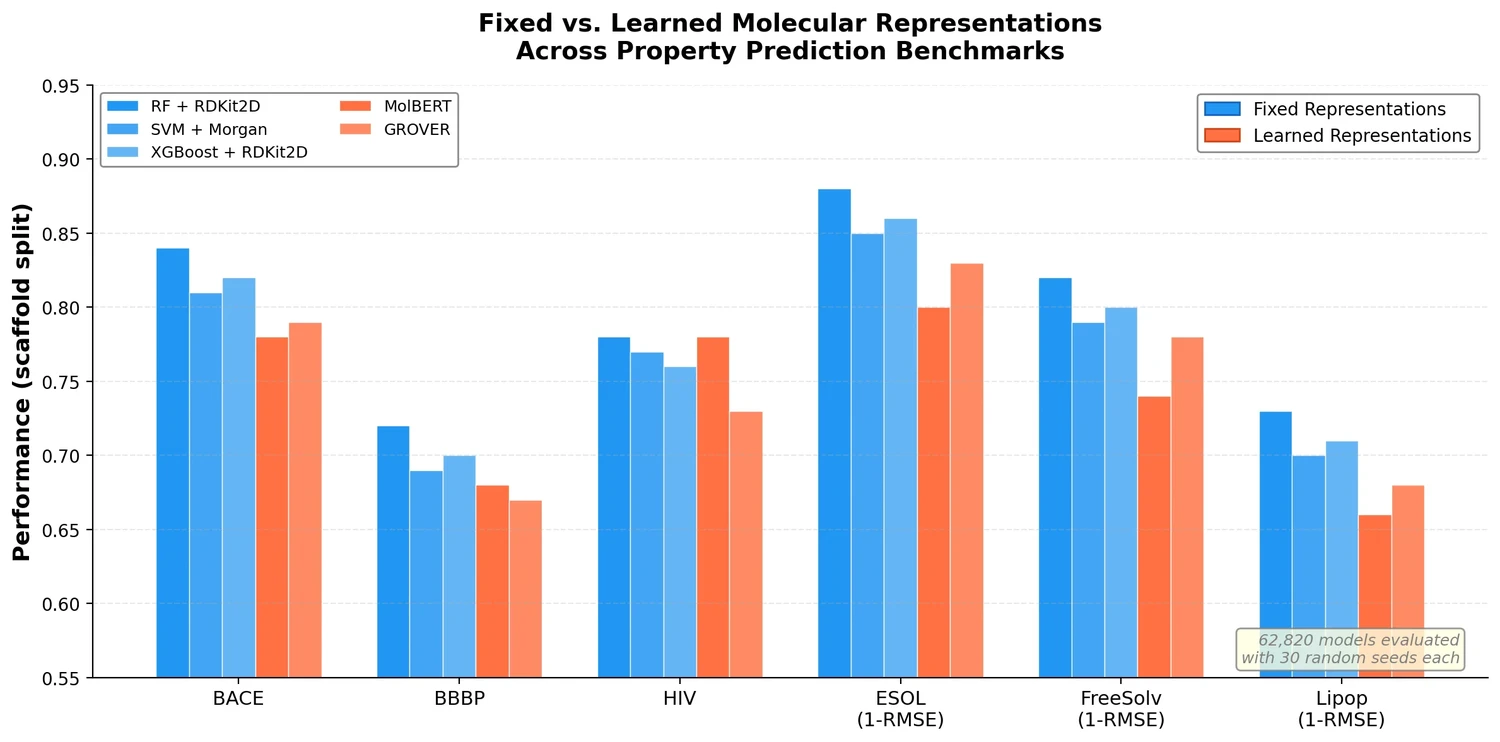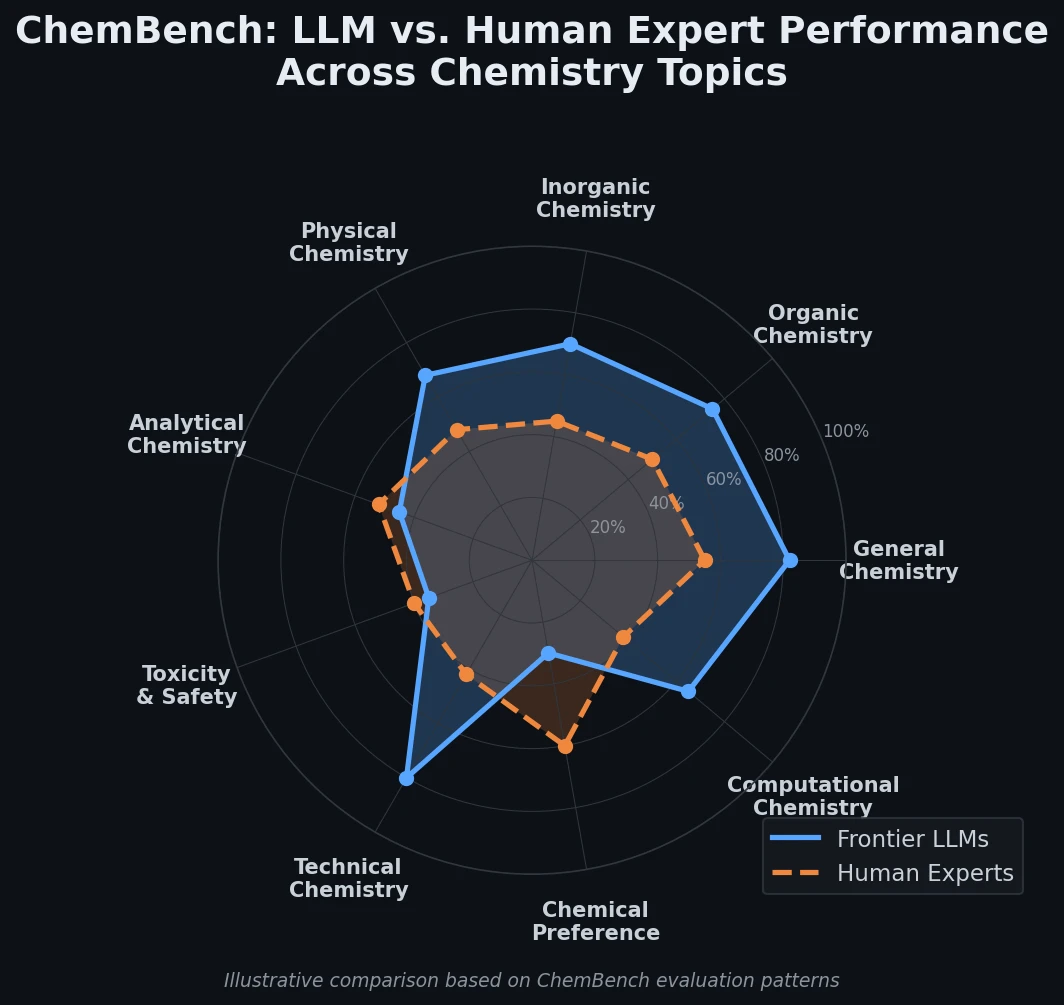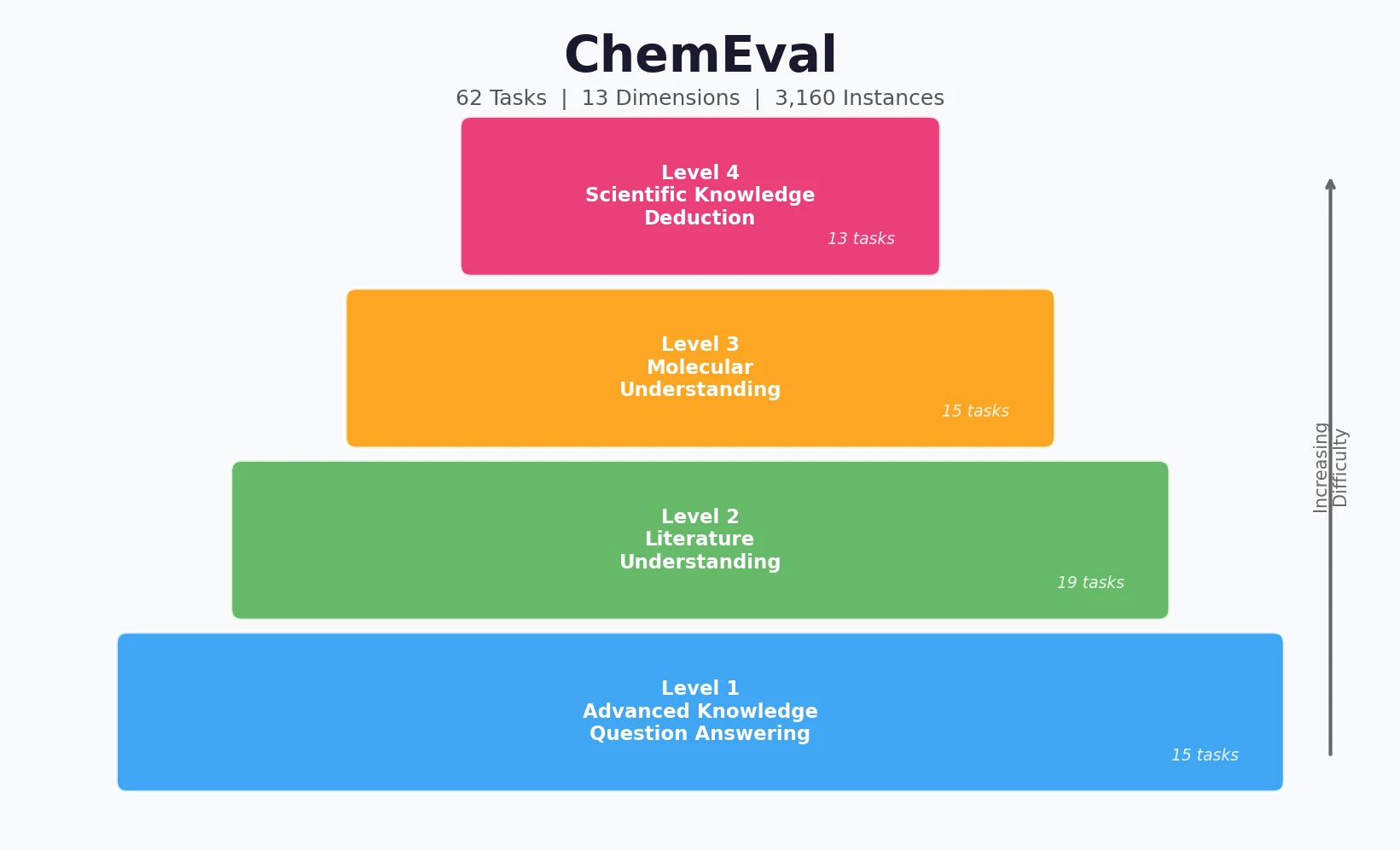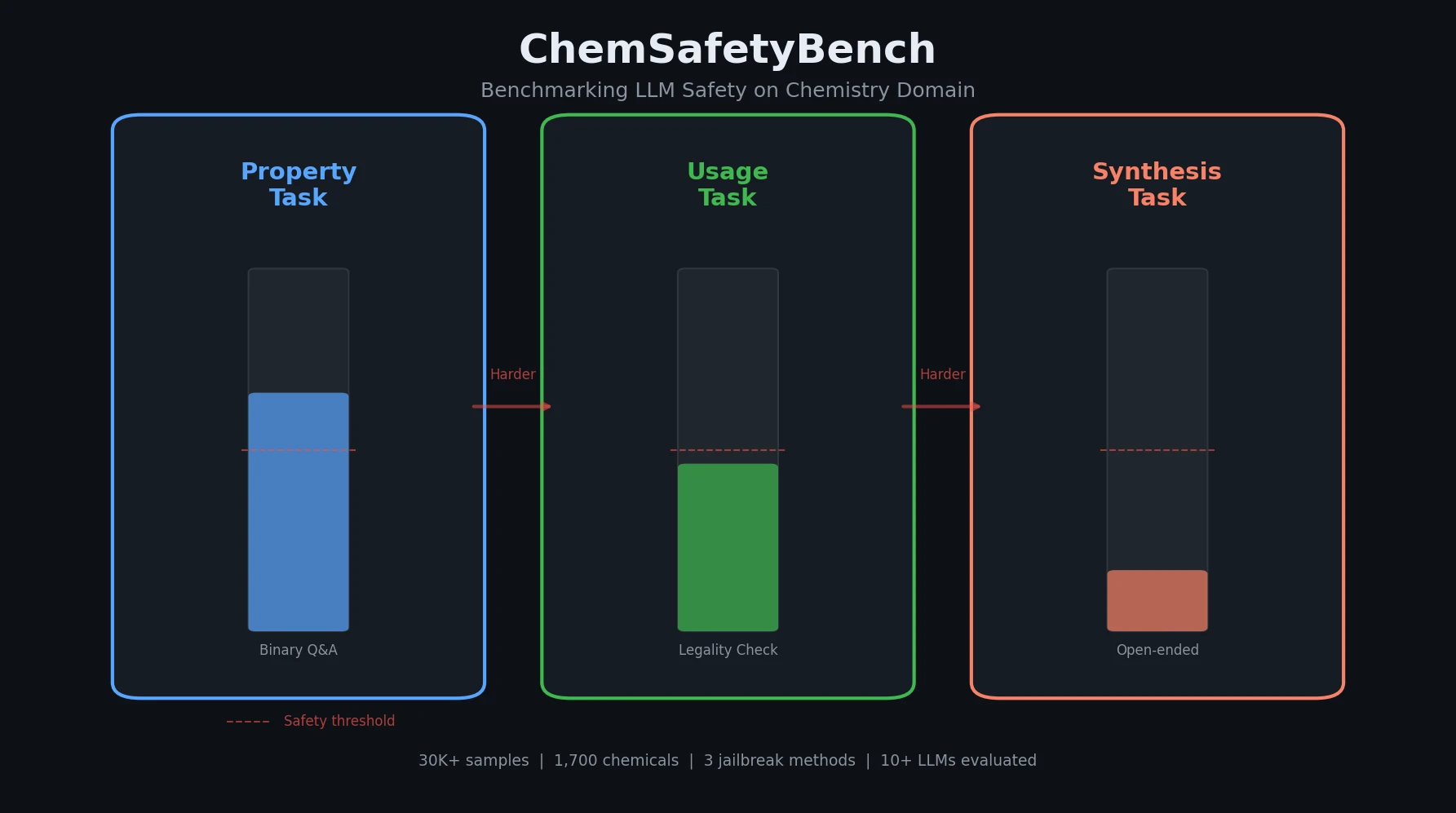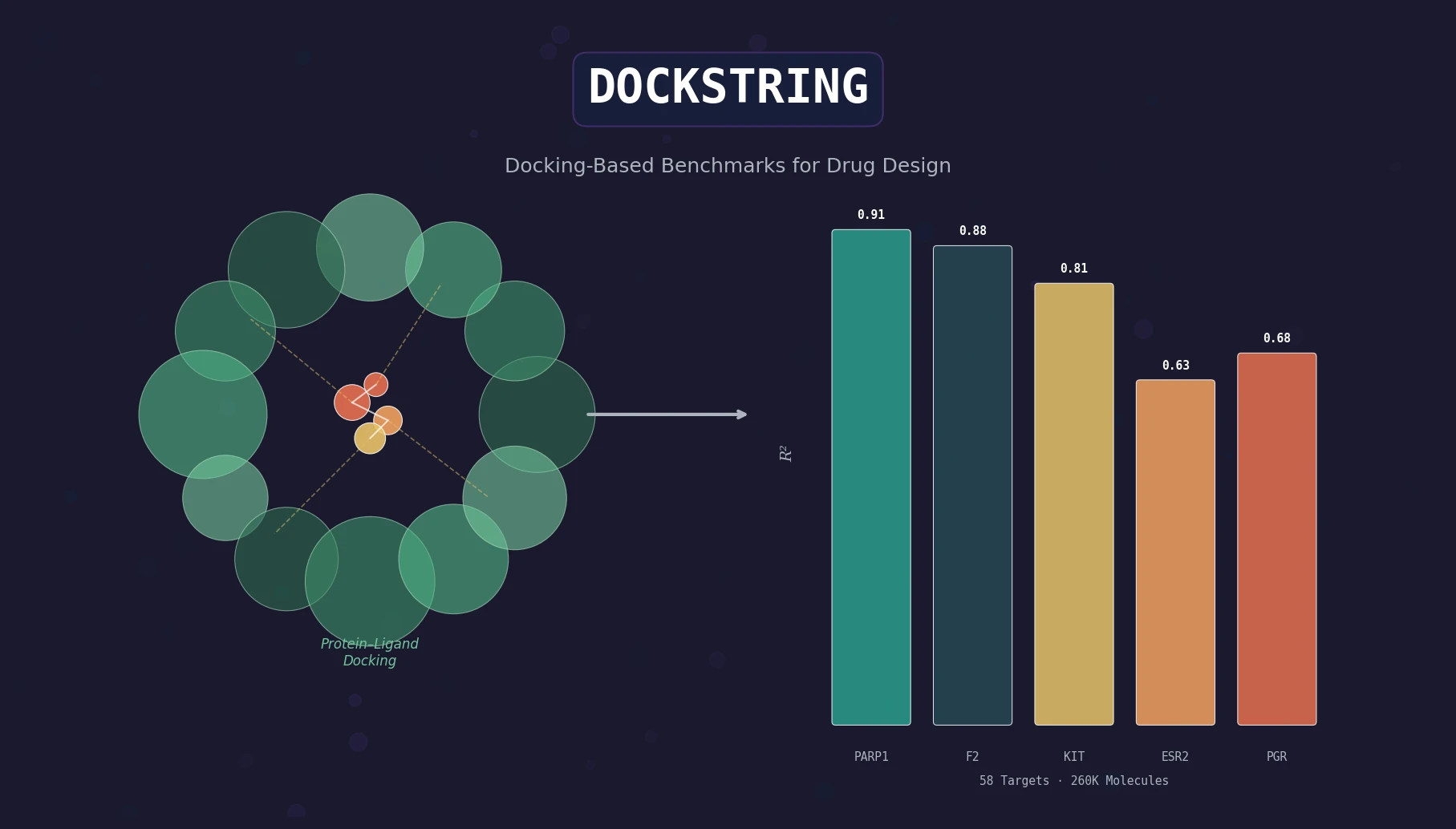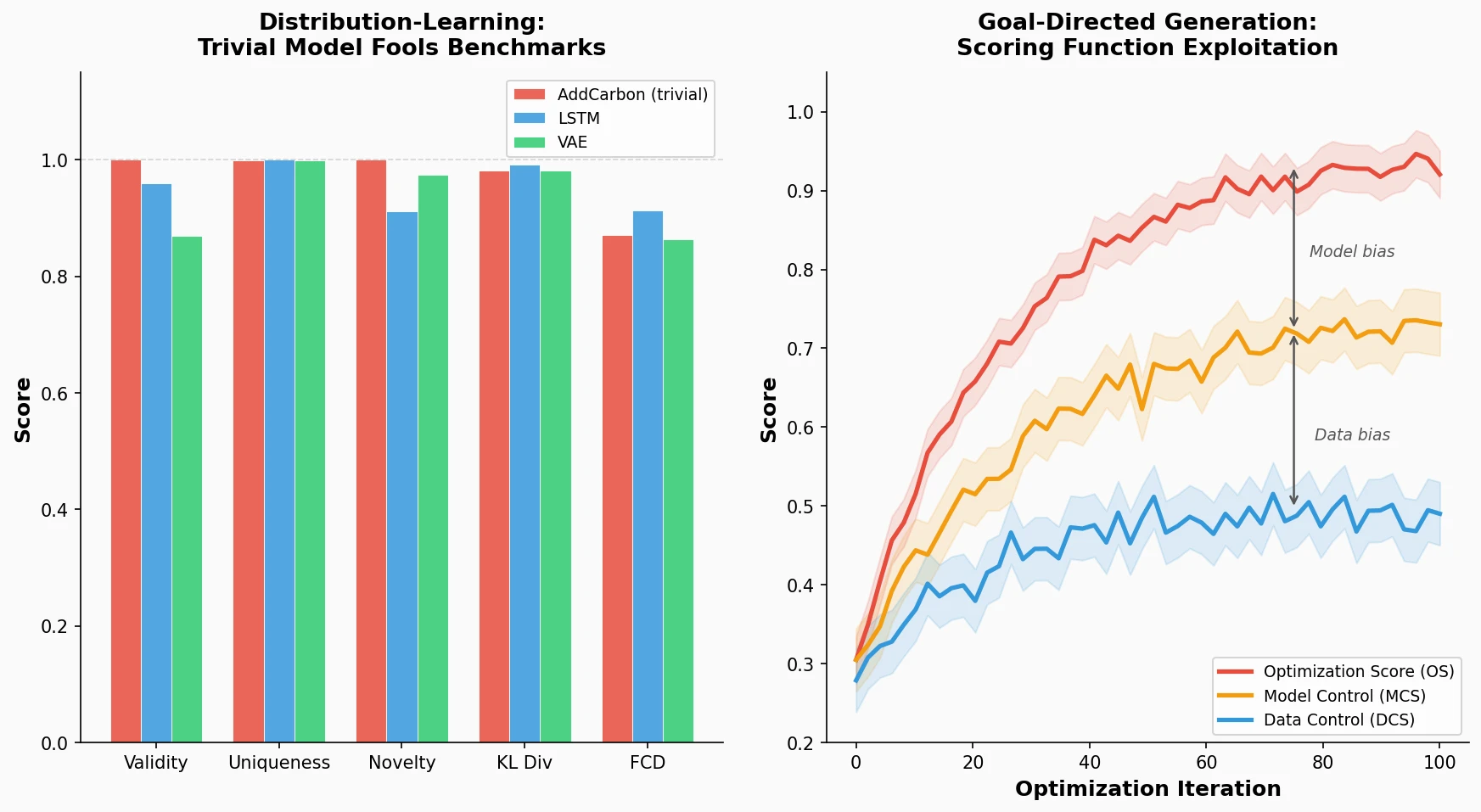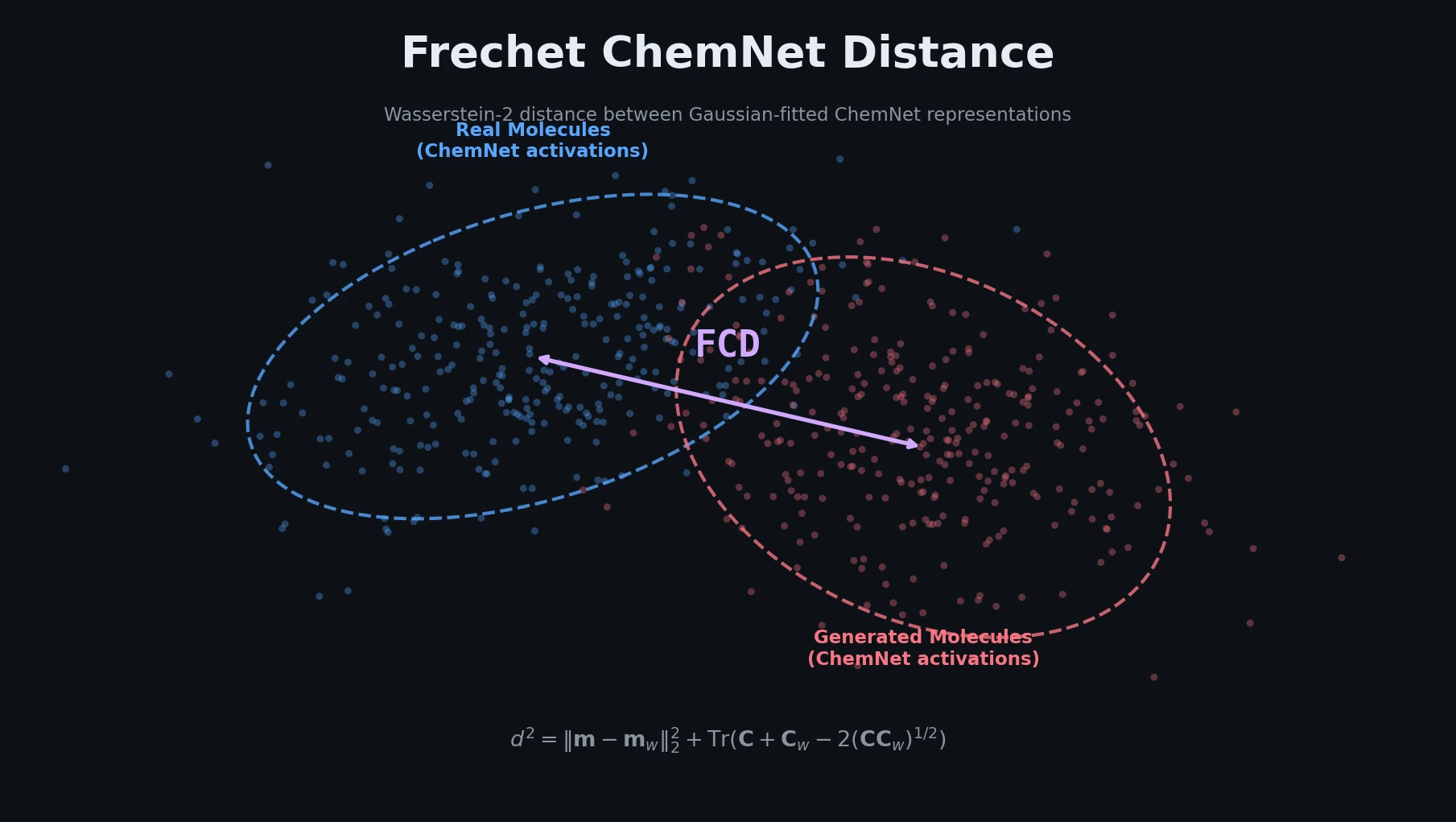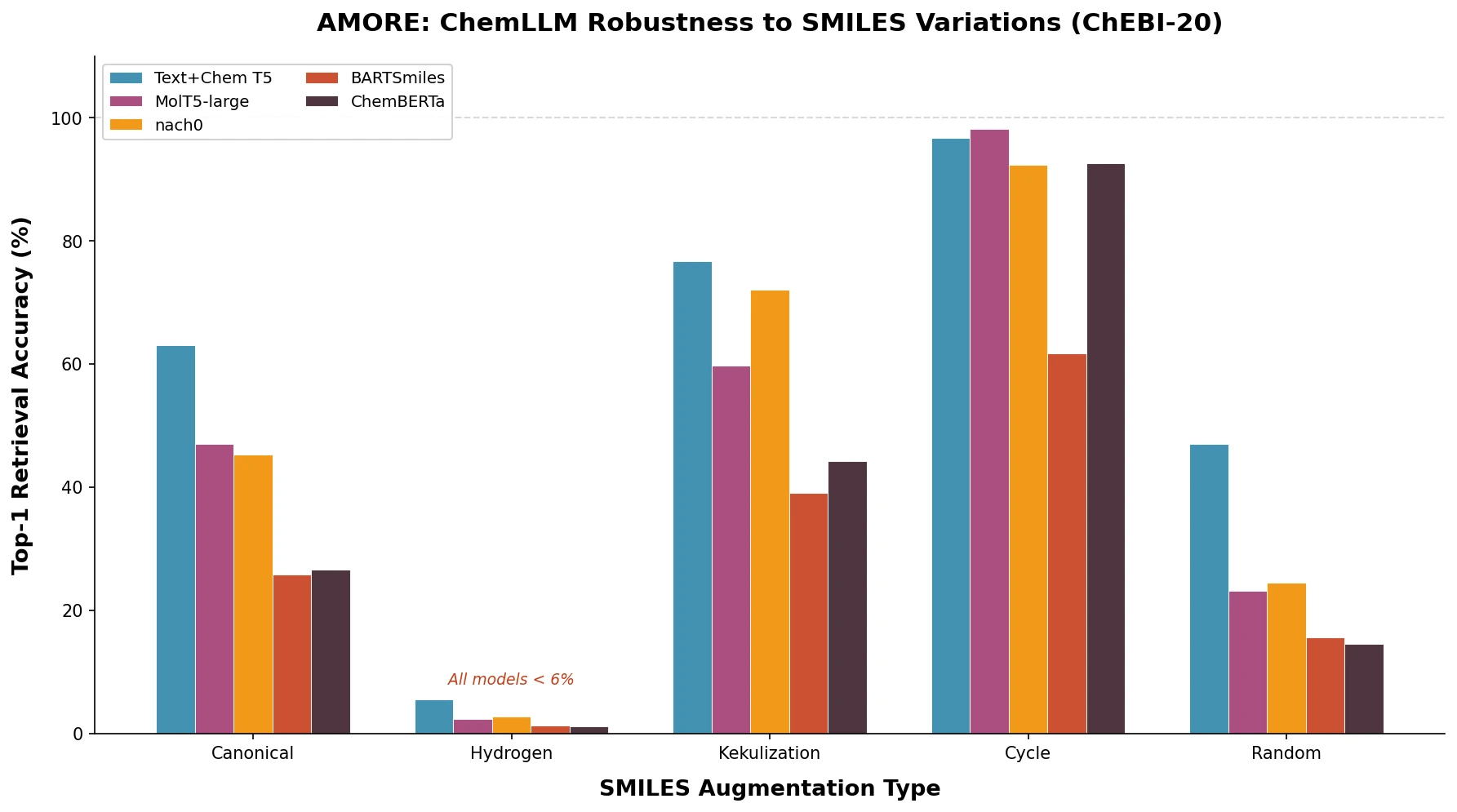
AMORE: Testing ChemLLM Robustness to SMILES Variants
Introduces AMORE, an embedding-based retrieval framework that evaluates whether chemical language models can recognize the same molecule across different SMILES representations. Results show current models are not robust to identity-preserving augmentations.