
GutenOCR: A Grounded Vision-Language Front-End for Documents
GutenOCR is a family of vision-language models designed to serve as a ‘grounded OCR front-end’, providing high-quality text transcription and explicit geometric grounding.
GutenOCR is a family of vision-language models designed to serve as a ‘grounded OCR front-end’, providing high-quality text transcription and explicit geometric grounding.
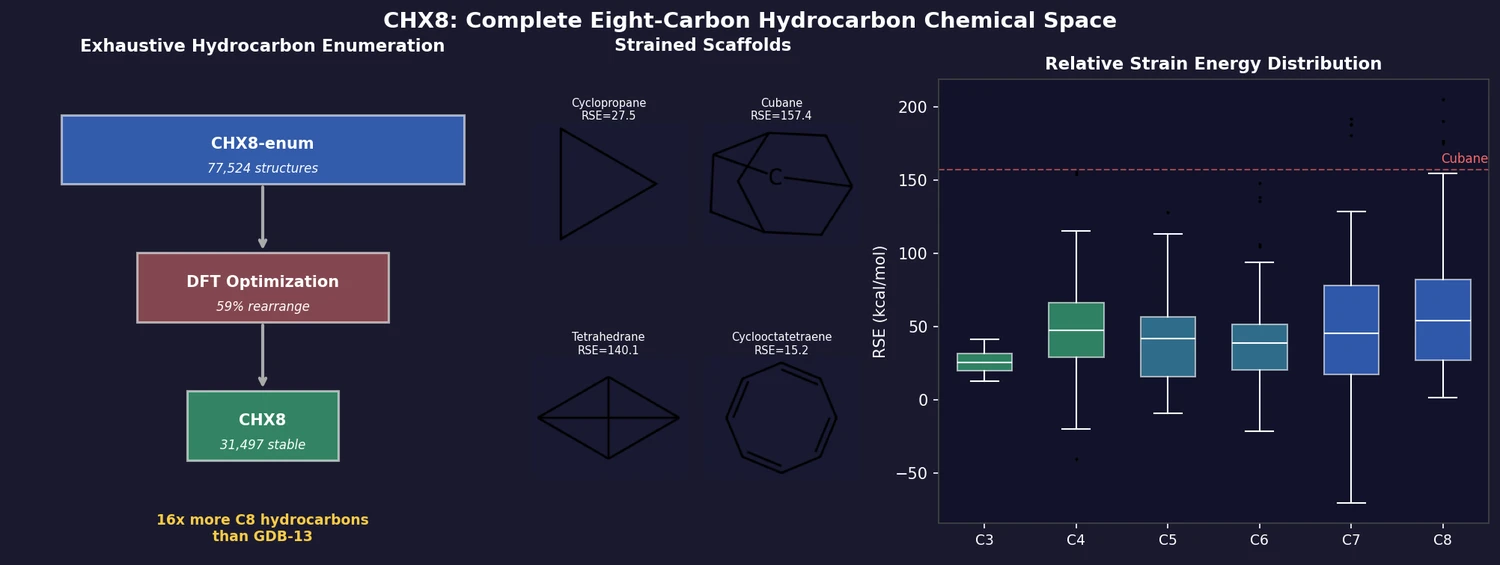
CHX8 exhaustively enumerates all mathematically feasible hydrocarbons with up to eight carbon atoms (77,524 structures), then DFT-optimizes them to identify 31,497 stable molecules. A universal relative strain energy (RSE) metric referenced to cyclohexane serves as a synthesizability proxy. CHX8 covers 16x more C8 hydrocarbons than GDB-13 and reveals that over 90% of novel structures should be synthetically accessible.
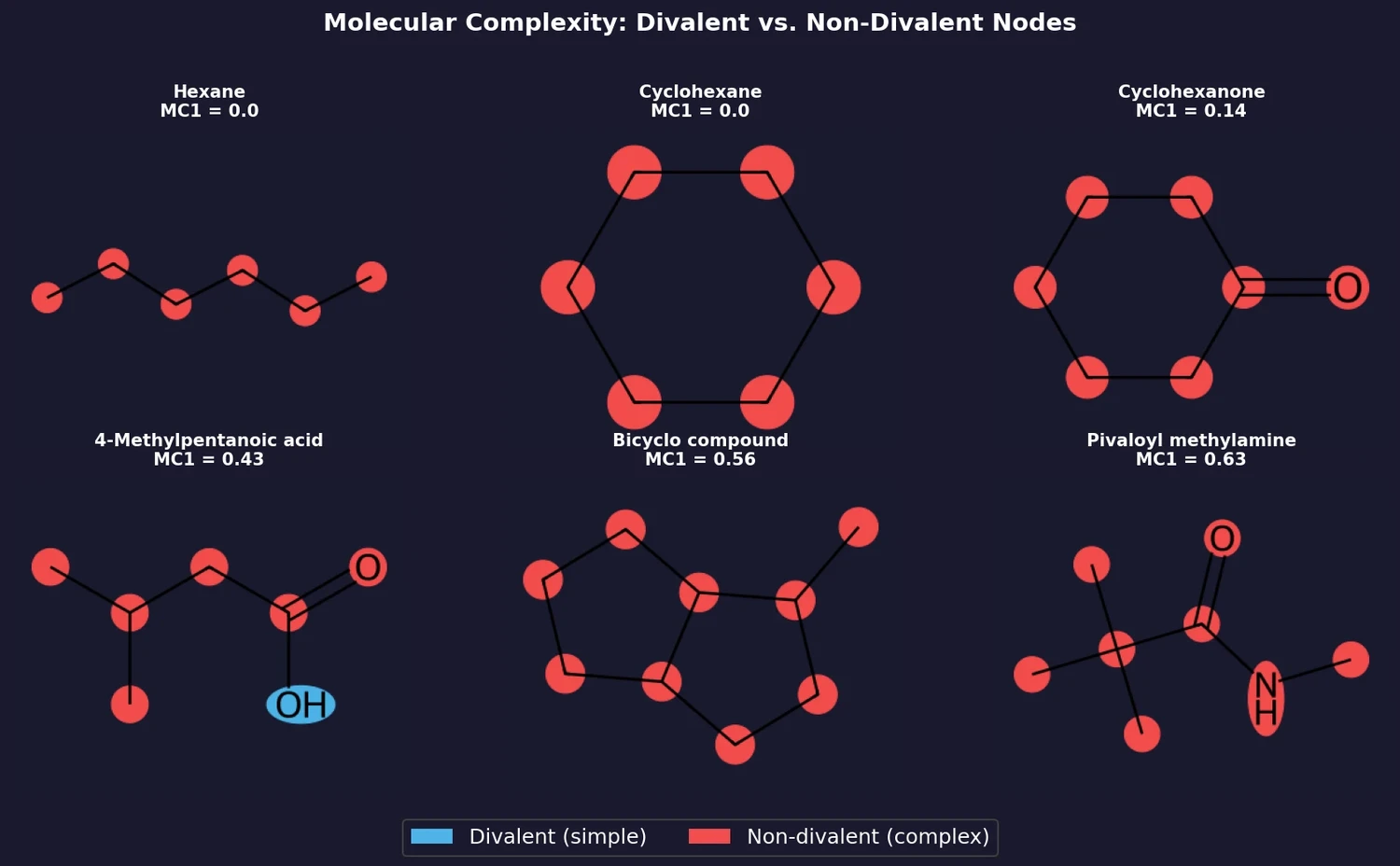
Buehler and Reymond introduce two molecular complexity measures, MC1 (fraction of non-divalent nodes) and MC2 (count of non-divalent nodes excluding carboxyl groups), derived from analyzing synthesizability patterns in GDB-enumerated molecules. They compare these measures against existing complexity scores across GDB-13s, ZINC, ChEMBL, and COCONUT.
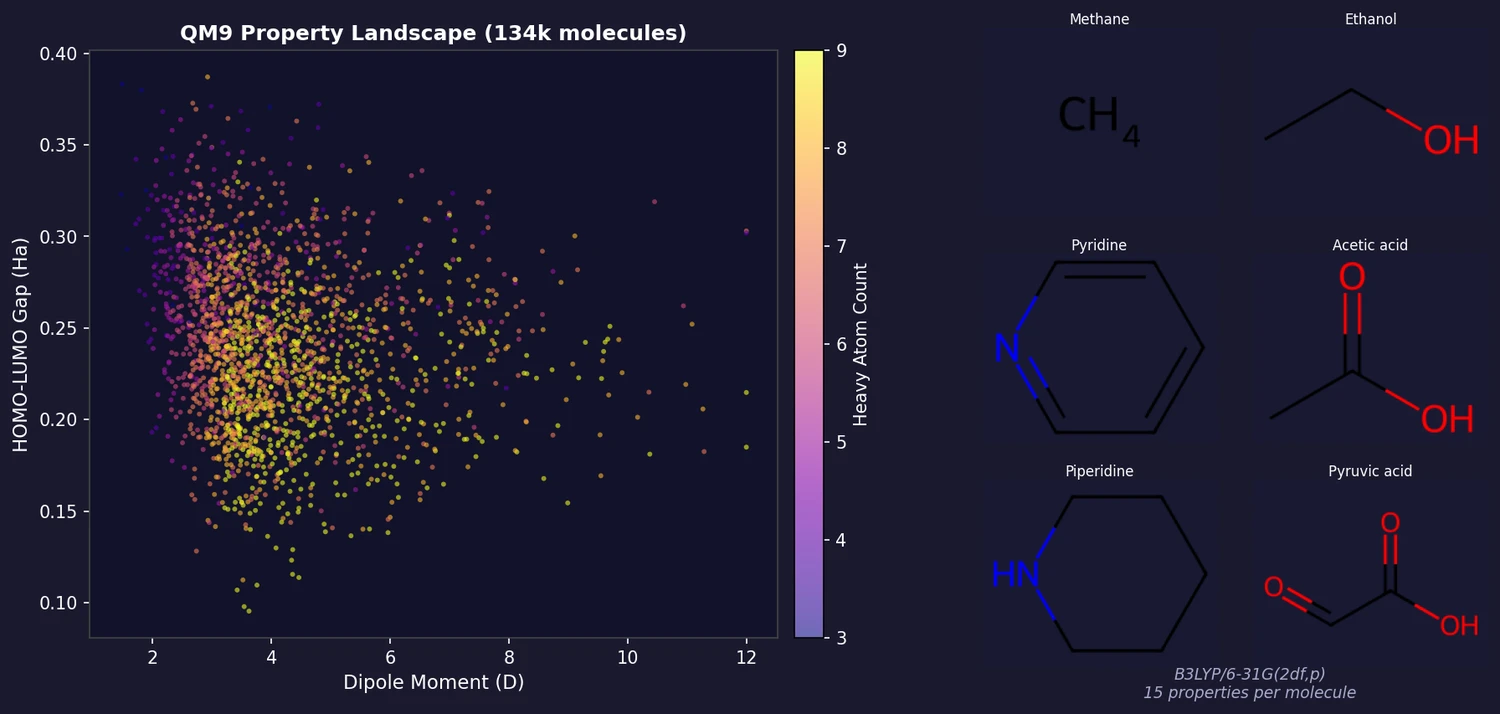
QM9 provides B3LYP/6-31G(2df,p)-level geometric, energetic, electronic, and thermodynamic properties for 133,885 small organic molecules (up to 9 heavy atoms of C, N, O, F) drawn from the GDB-17 chemical universe. It is one of the most widely used benchmarks in molecular machine learning.
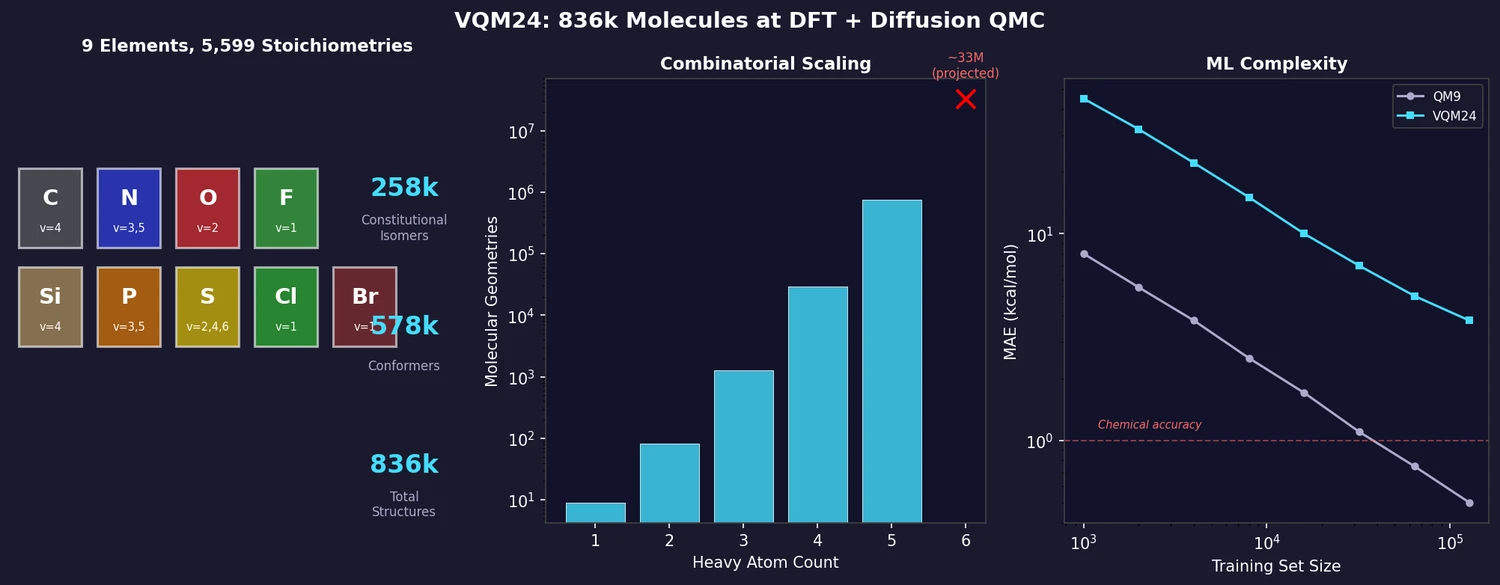
VQM24 exhaustively enumerates all neutral closed-shell molecules with up to 5 heavy atoms from C, N, O, F, Si, P, S, Cl, Br, yielding 258k constitutional isomers and 578k conformers (836k total). Properties are computed at the wB97X-D3/cc-pVDZ level, with diffusion QMC energies for 10,793 molecules up to 4 heavy atoms. ML models show up to 8x higher errors than on QM9, making VQM24 a more challenging benchmark.
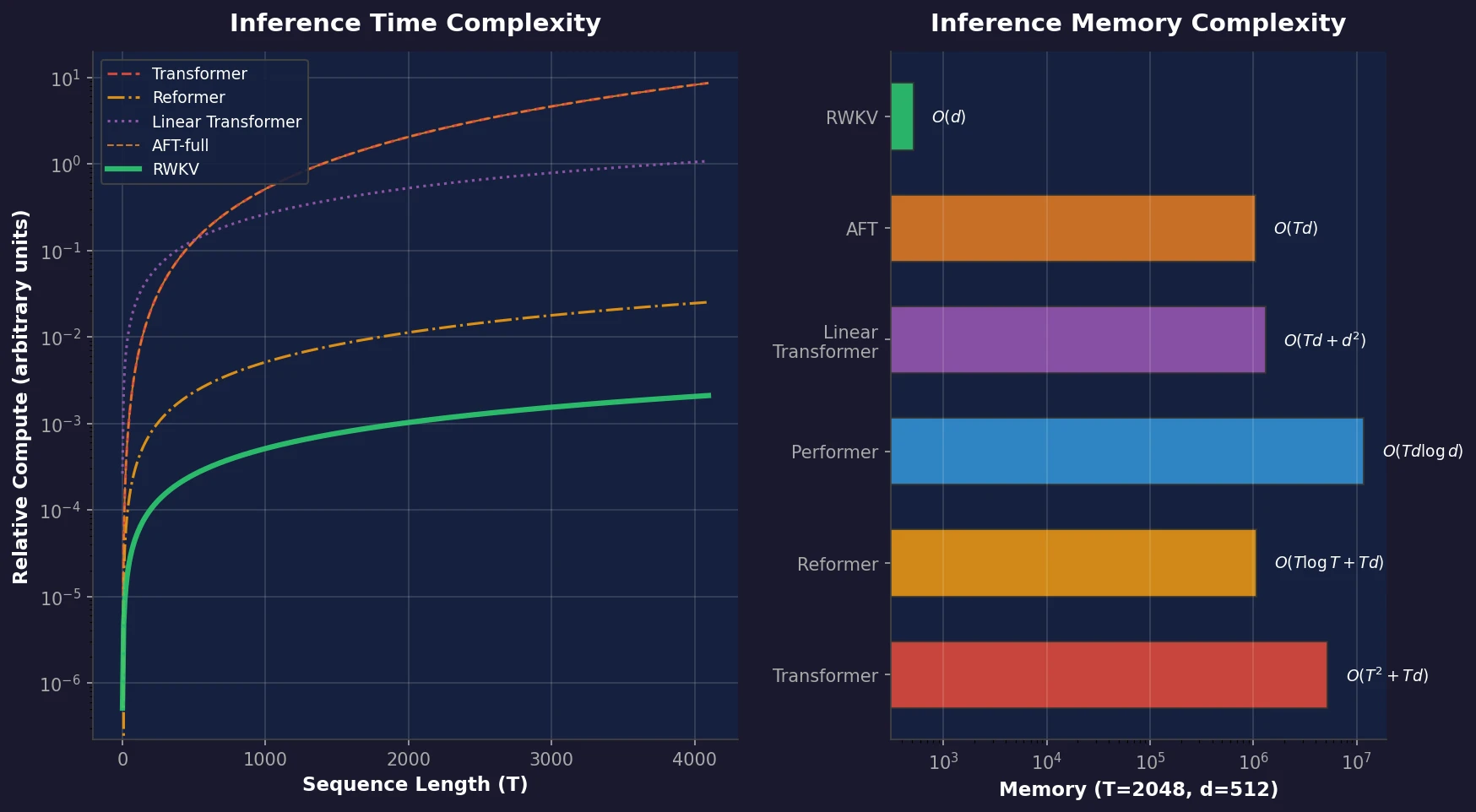
RWKV is a novel sequence model that achieves transformer-level performance while maintaining linear time and constant memory complexity during inference, scaled up to 14 billion parameters.
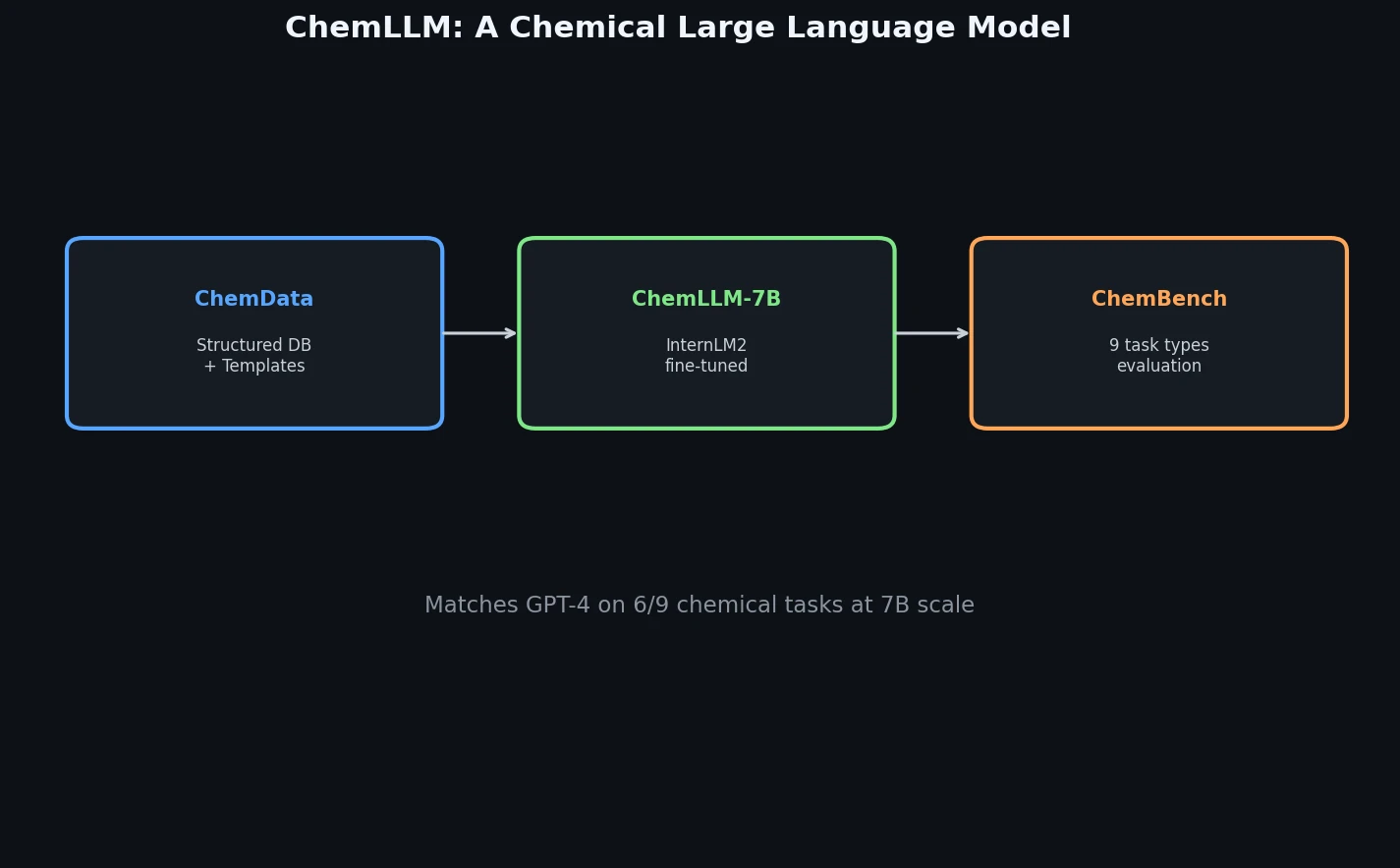
ChemLLM presents a comprehensive framework for chemistry-specific language modeling, including a 7M-sample instruction tuning dataset (ChemData), a 4,100-question benchmark (ChemBench), and a two-stage fine-tuned model that matches GPT-4 on core chemical tasks.
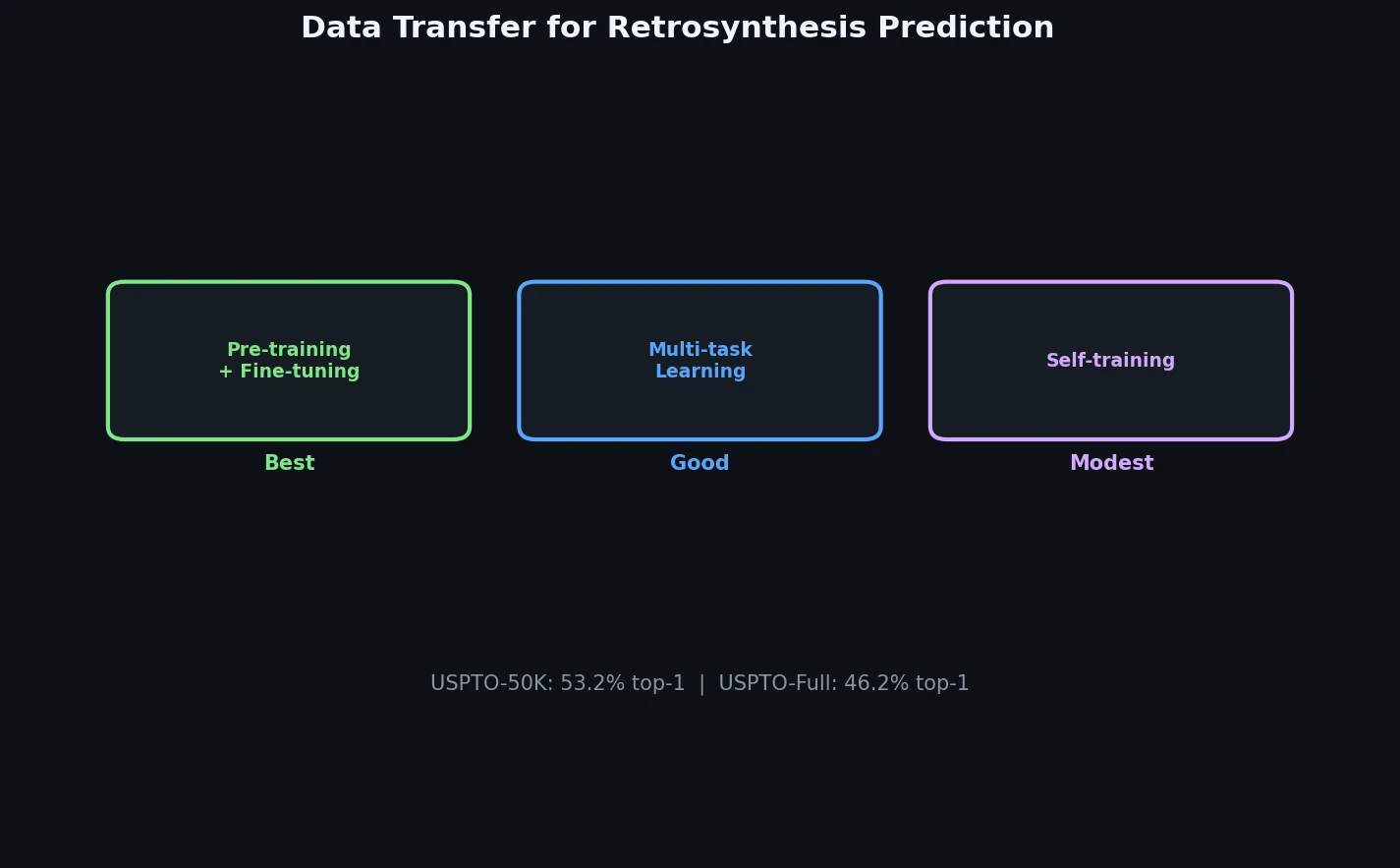
A systematic study of data transfer techniques (joint training, self-training, pre-training plus fine-tuning) applied to Transformer-based retrosynthesis. Pre-training on USPTO-Full followed by fine-tuning on USPTO-50K achieves the best results, improving top-1 accuracy from 35.3% to 57.4%.
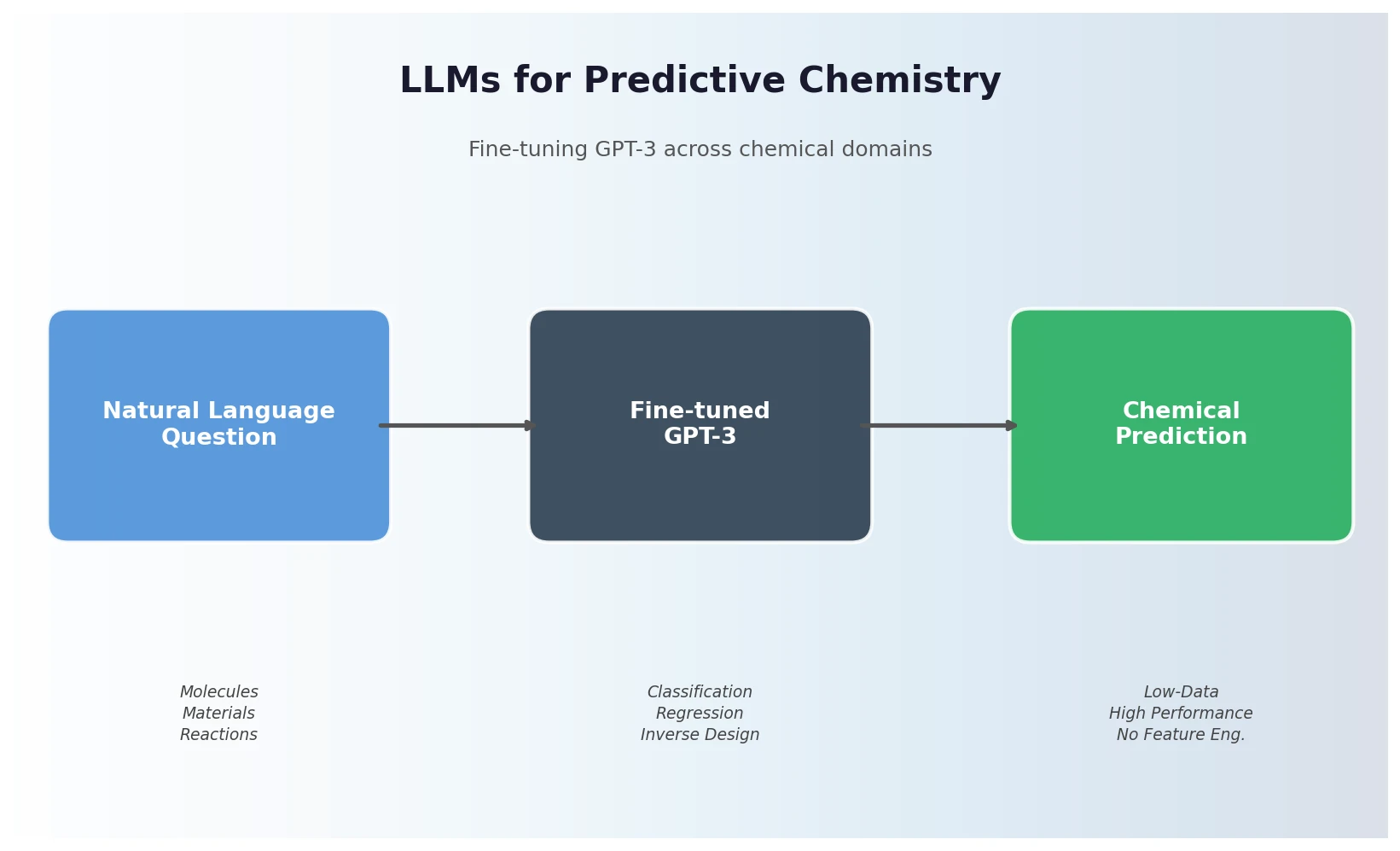
Jablonka et al. show that fine-tuning GPT-3 on natural language chemistry questions achieves competitive or superior performance to dedicated ML models across 15 benchmarks, with particular strength in low-data settings and inverse molecular design.
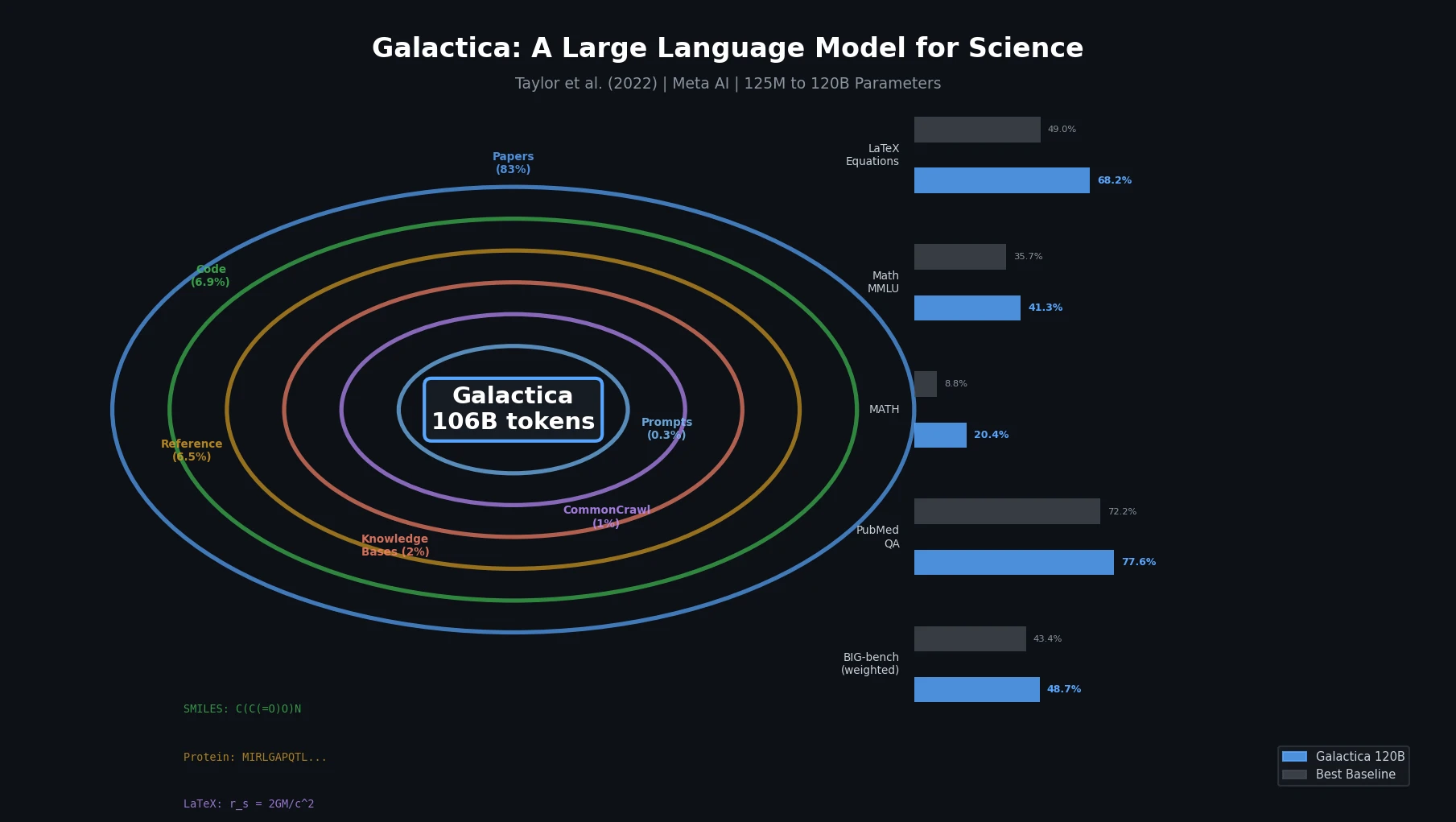
Galactica trains a decoder-only Transformer on a curated 106B-token scientific corpus spanning papers, proteins, and molecules, achieving strong results on scientific QA, mathematical reasoning, and citation prediction.
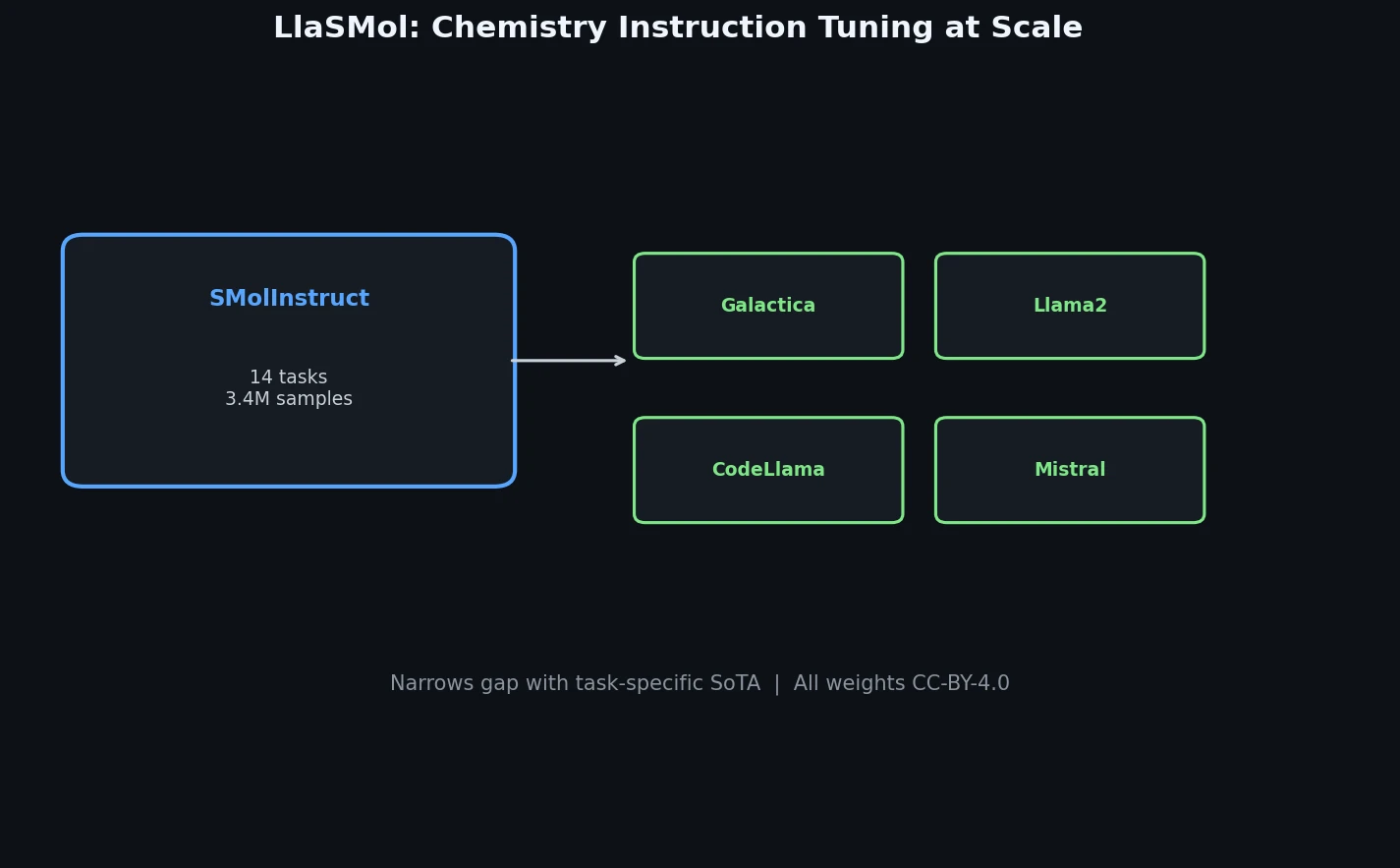
LlaSMol fine-tunes Mistral, Llama 2, and other open-source LLMs on SMolInstruct, a 3.3M-sample instruction tuning dataset covering 14 chemistry tasks. The Mistral-based model outperforms GPT-4 and Claude 3 Opus across all tasks.

PharmaGPT is a suite of domain-specific LLMs (13B and 70B parameters) built on LLaMA with continued pretraining on biopharmaceutical and chemical data, achieving strong results on NAPLEX and Chinese pharmacist exams.