Overview
This project provides a complete “input-to-analysis” workflow for simulating adatom diffusion on FCC metal surfaces. It demonstrates how to set up surface diffusion simulations in LAMMPS, manage EAM potentials, and automatically parse trajectory data into publication-ready visualizations using Python.
The workflow covers two material systems (Copper (Cu) and Platinum (Pt)) providing comparative datasets that highlight how atomic mass and bonding strength affect surface dynamics.
Features
Simulation Architecture
The project separates simulation logic from analysis code:
| Directory | Description |
|---|---|
/adatom_cu | Copper adatom diffusion on Cu(100) |
/adatom_pt | Platinum adatom diffusion on Pt(100) |
Each directory contains:
- LAMMPS input scripts (
.infiles) defining the physics - EAM potential files for accurate metallic bonding
- Python analysis scripts for trajectory and energy parsing
Key Features
- EAM Potentials: Uses Embedded Atom Method alloy potentials to accurately model metallic bonding and surface energies, providing accuracy beyond simple Lennard-Jones potentials
- Automated Analysis: Python pipeline (
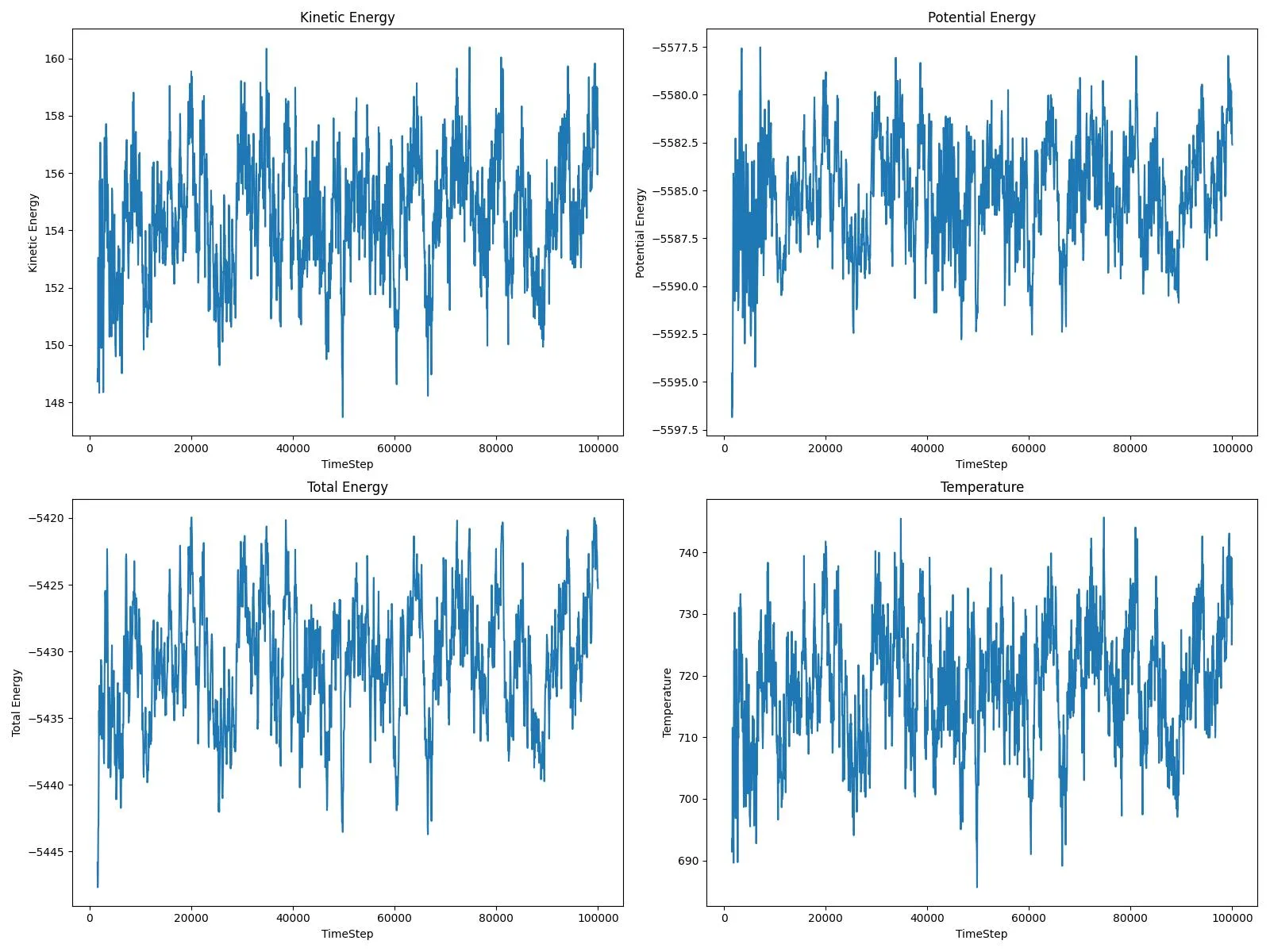
plot_energy.py,plot_xy.py) that parses raw thermodynamic logs and trajectory dumps to generate “health check” dashboards - Workflow Orchestration: Demonstrates the “Input → Simulation → Analysis” loop, automating the transition from raw
.lammpstrjfiles to publication-ready plots - Kokkos Support: Includes high-performance execution commands for GPU/multi-threaded runs
Simulation Parameters
| Parameter | Value | Purpose |
|---|---|---|
| Ensemble | NVT → NVE | Equilibration followed by energy conservation checks |
| Potential | EAM/alloy | Accurate metallic bonding for surface dynamics |
| Minimization | CG (1.0e-4) | Remove steric overlaps before dynamics |
| Output | 5 fs resolution | High-fidelity trajectory capture |
Usage
The repository includes LAMMPS input scripts and Python analysis scripts. Run the LAMMPS scripts to generate trajectory data, then use the Python scripts to visualize the results.
Results
This workflow is documented in detail in companion blog posts:
- LAMMPS Tutorial: Copper and Platinum Adatom Diffusion - Complete setup walkthrough with line-by-line script explanation and comparison of how heavier atoms behave differently on surfaces