Transformers for Molecular Property Prediction Review
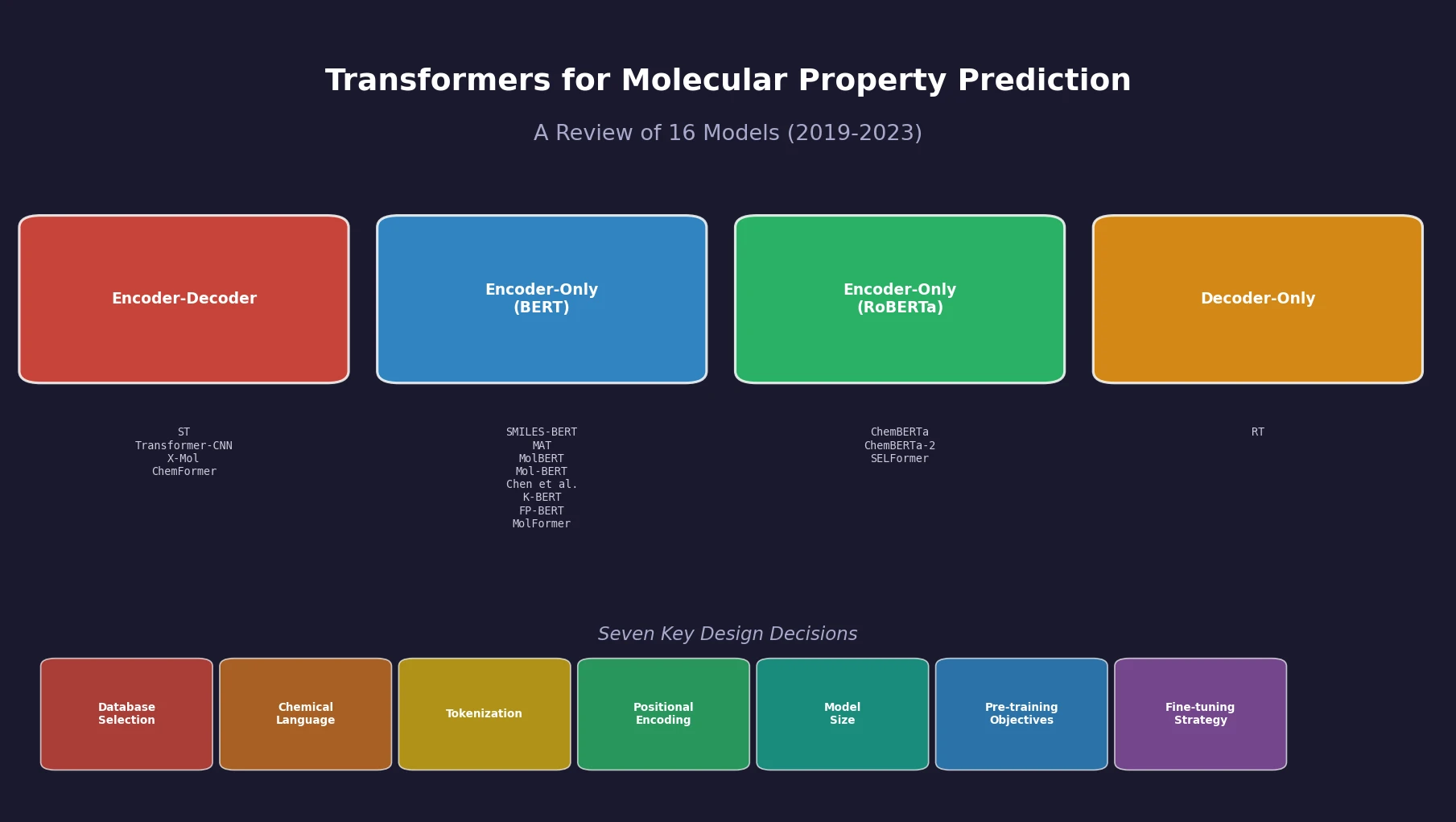
Sultan et al. review 16 sequence-based transformer models for molecular property prediction, systematically analyzing seven design decisions (database selection, chemical language, tokenization, positional encoding, model size, pre-training objectives, and fine-tuning strategy) and identifying a critical need for standardized evaluation practices.