GuacaMol: Benchmarking Models for De Novo Molecular Design
GuacaMol provides an open-source benchmarking framework with 5 distribution-learning and 20 goal-directed tasks to standardize evaluation of de novo molecular design models.
GuacaMol provides an open-source benchmarking framework with 5 distribution-learning and 20 goal-directed tasks to standardize evaluation of de novo molecular design models.
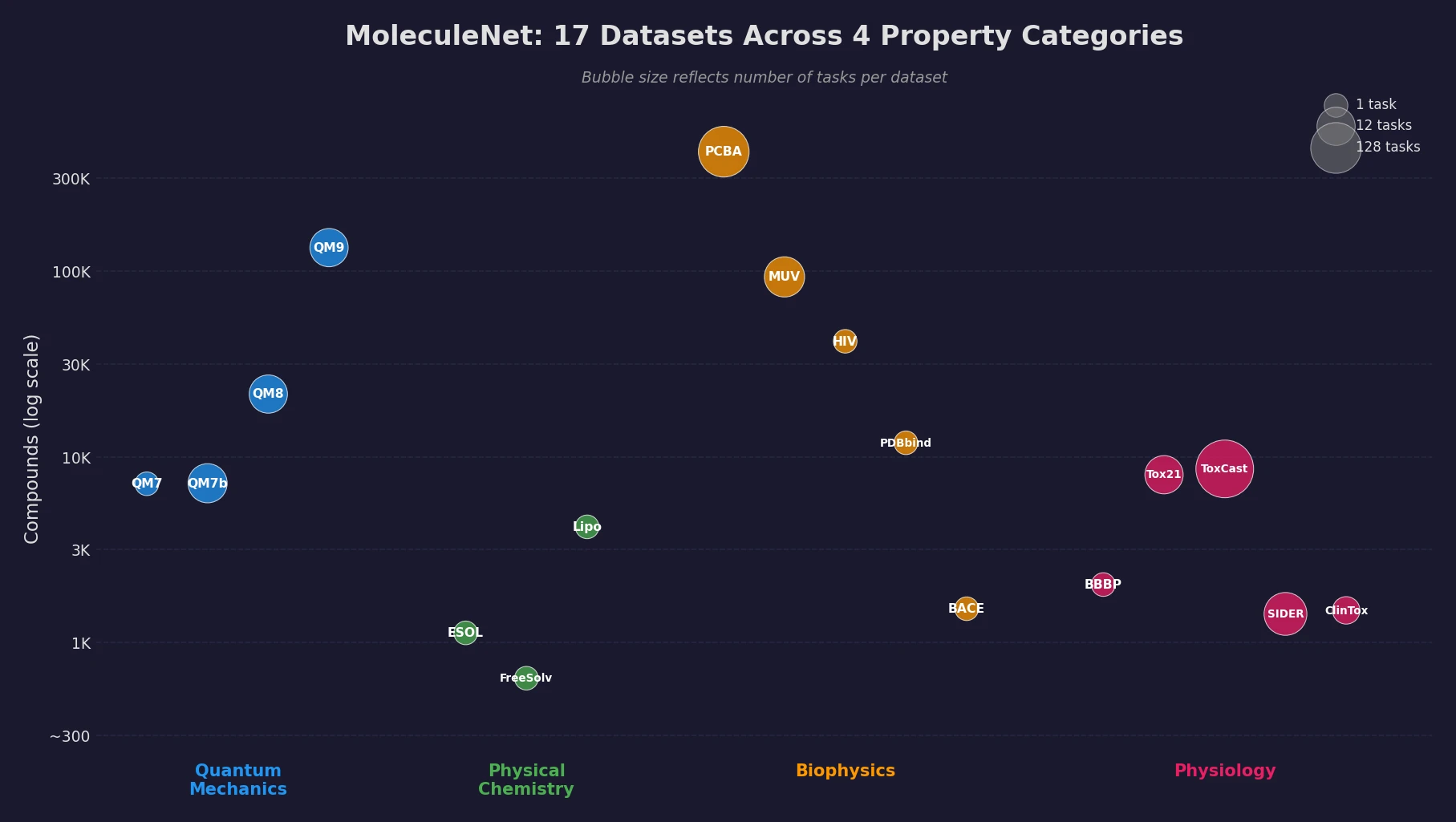
MoleculeNet introduces a large-scale benchmark suite for molecular machine learning, curating over 700,000 compounds across 17 datasets with standardized metrics, data splits, and featurization methods integrated into the DeepChem open-source library.
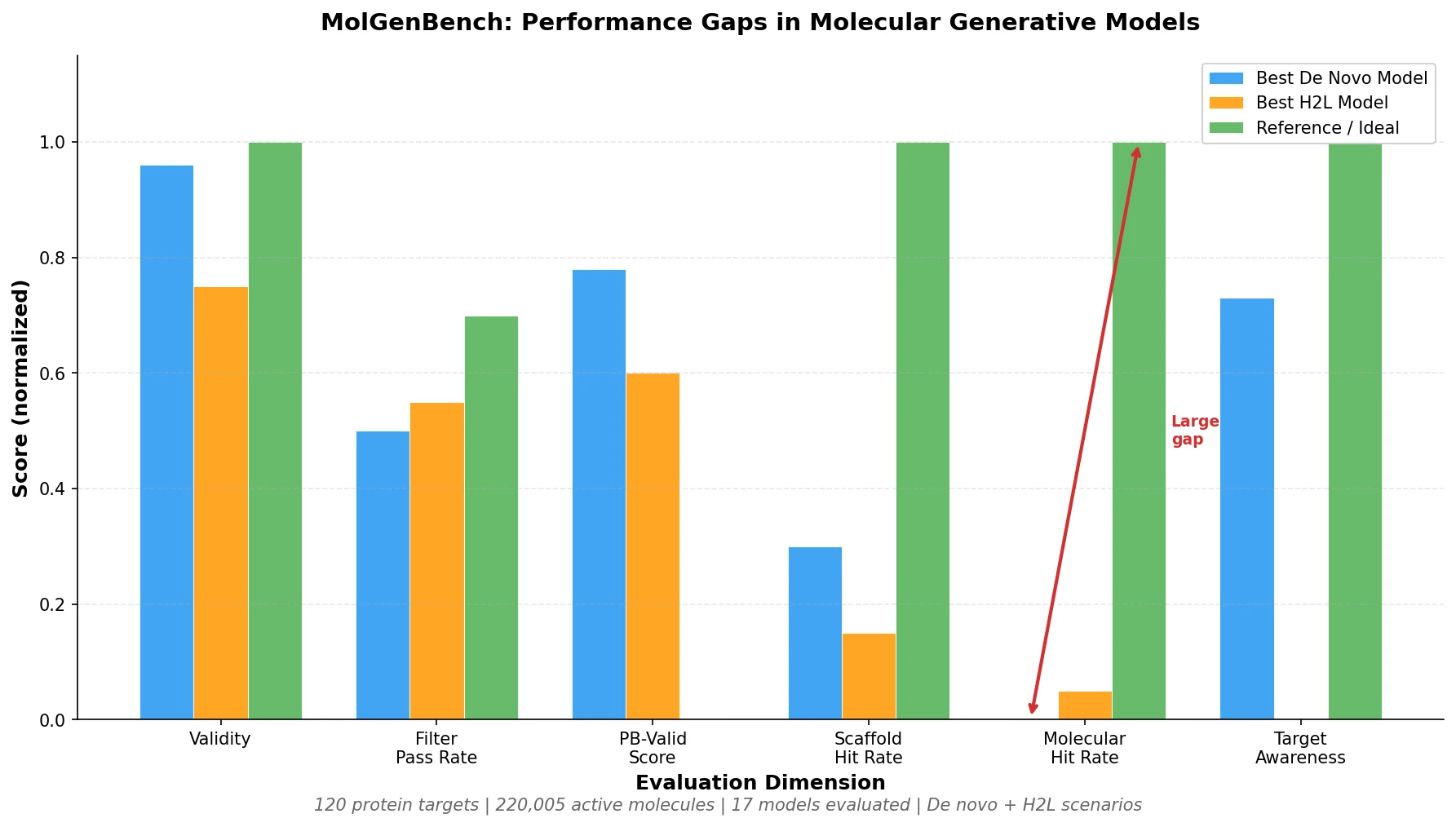
MolGenBench introduces a comprehensive benchmark for evaluating molecular generative models in realistic drug discovery settings, spanning de novo design and hit-to-lead optimization across 120 protein targets with 220,005 experimentally validated actives.
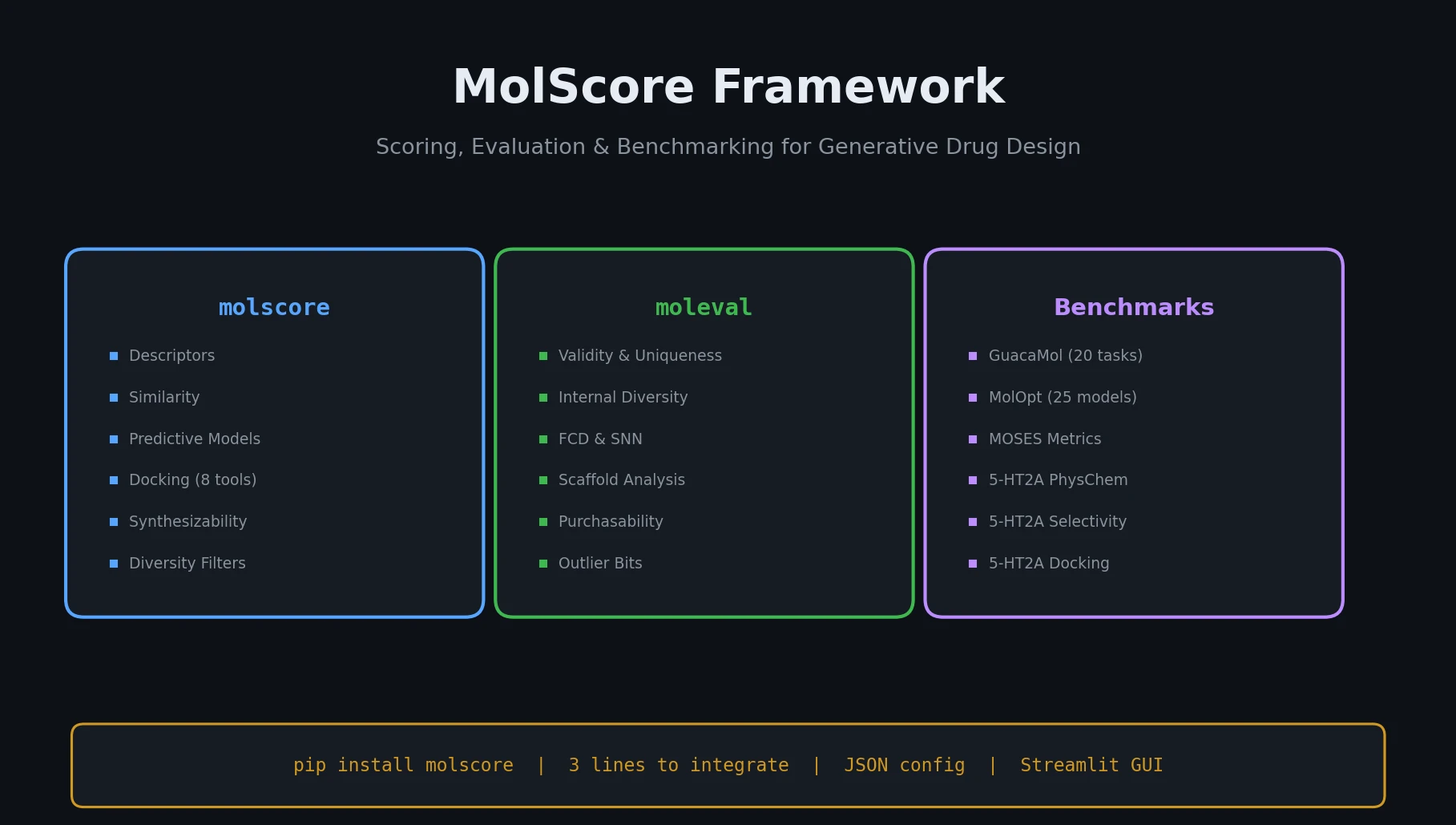
MolScore is an open-source framework that unifies scoring functions, evaluation metrics, and benchmarks for generative molecular design, with configurable objectives and GUI support.
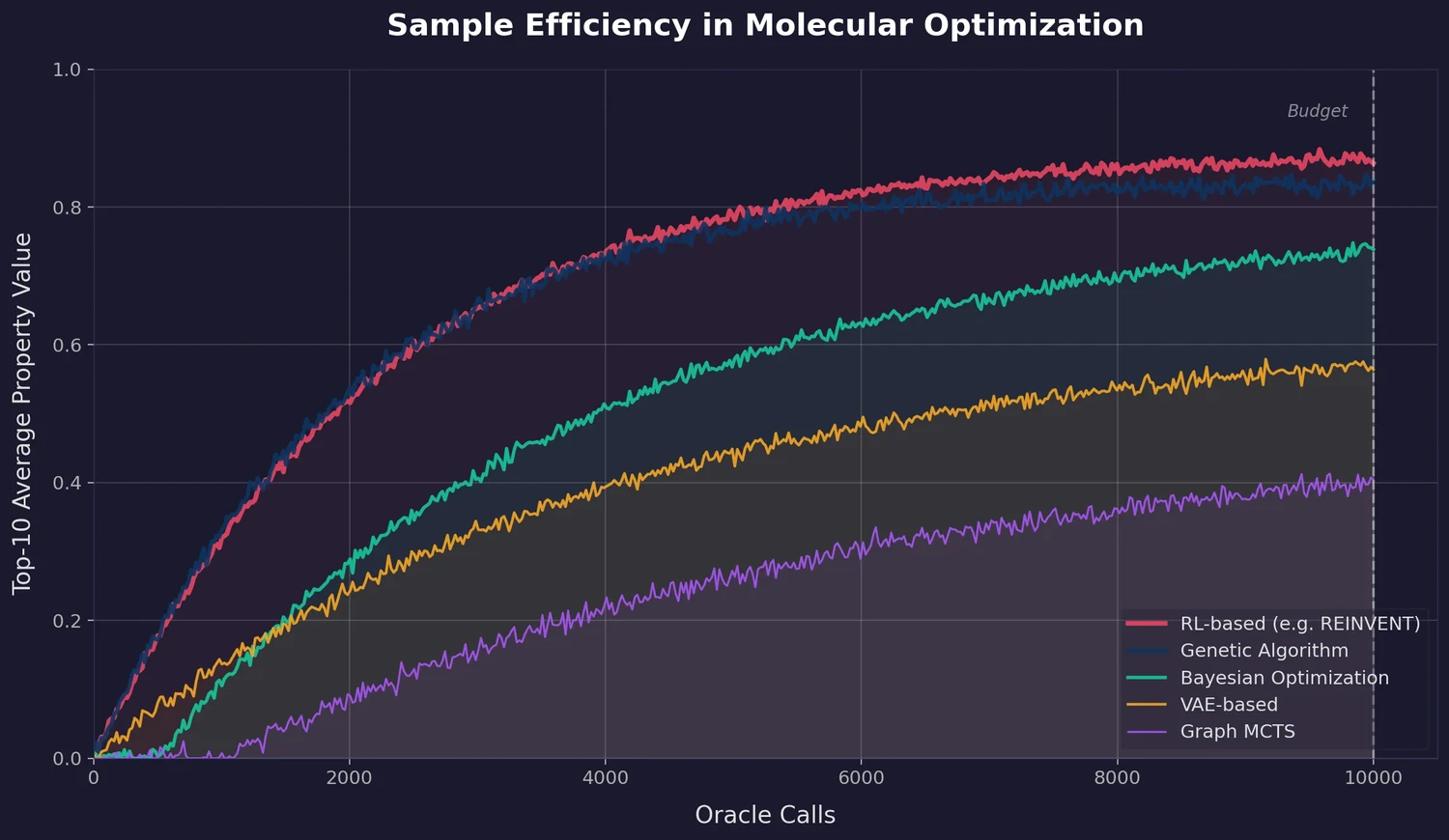
A large-scale benchmark of 25 molecular optimization methods on 23 oracles under constrained oracle budgets, showing that sample efficiency is a critical and often neglected dimension of evaluation.
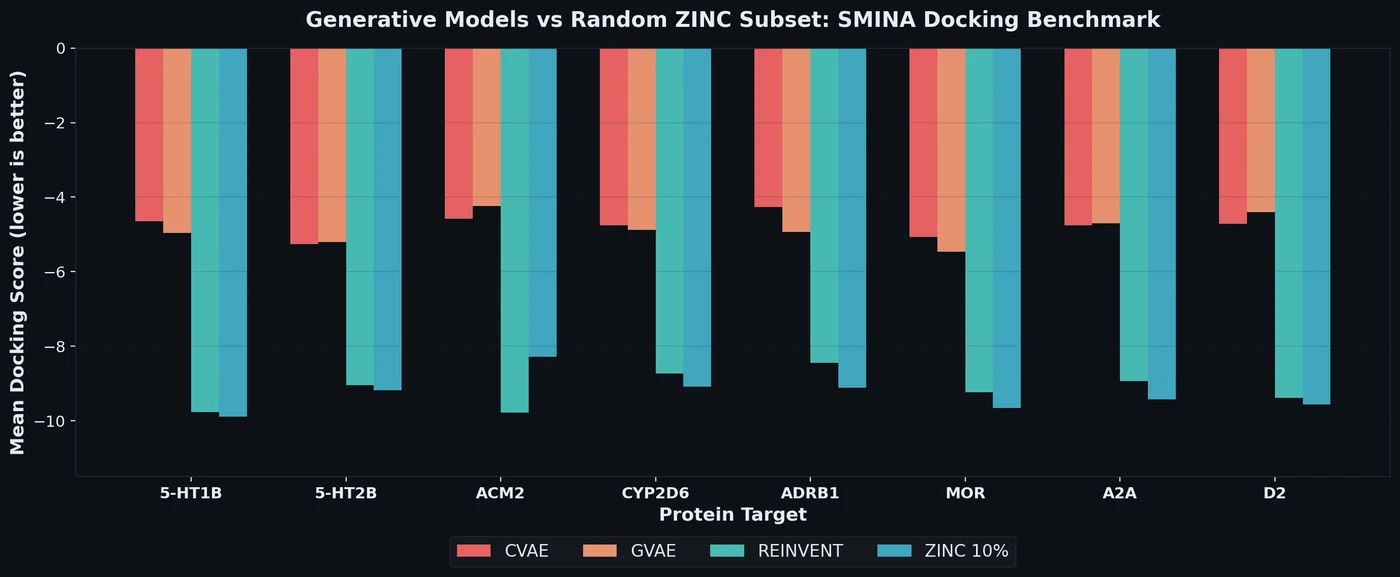
Proposes a benchmark for de novo drug design using SMINA docking scores across eight drug targets, revealing that popular generative models fail to outperform random ZINC subsets.
Tartarus introduces a modular suite of realistic molecular design benchmarks grounded in computational chemistry simulations. Benchmarking eight generative models reveals that no single algorithm dominates all tasks, and simple genetic algorithms often outperform deep generative models.
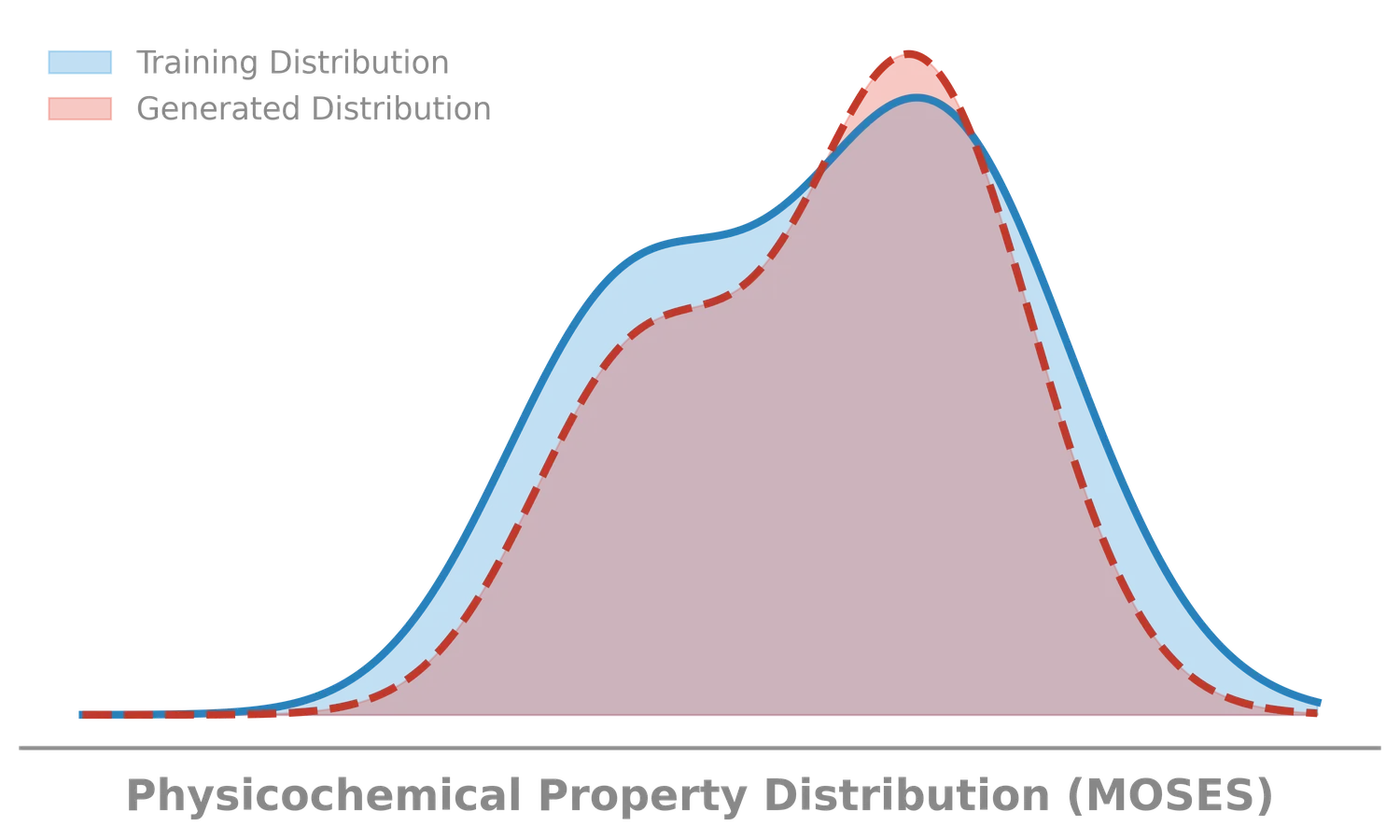
MOSES introduces a comprehensive benchmarking platform for molecular generative models, offering standardized datasets, evaluation metrics, and baselines. By providing a unified measuring stick, it aims to resolve reproducibility challenges in chemical distribution learning.

ChemBERTa-3 provides a unified, scalable infrastructure for pretraining and benchmarking chemical foundation models. It addresses reproducibility gaps in previous studies like MoLFormer through standardized scaffold splitting and open-source tooling.
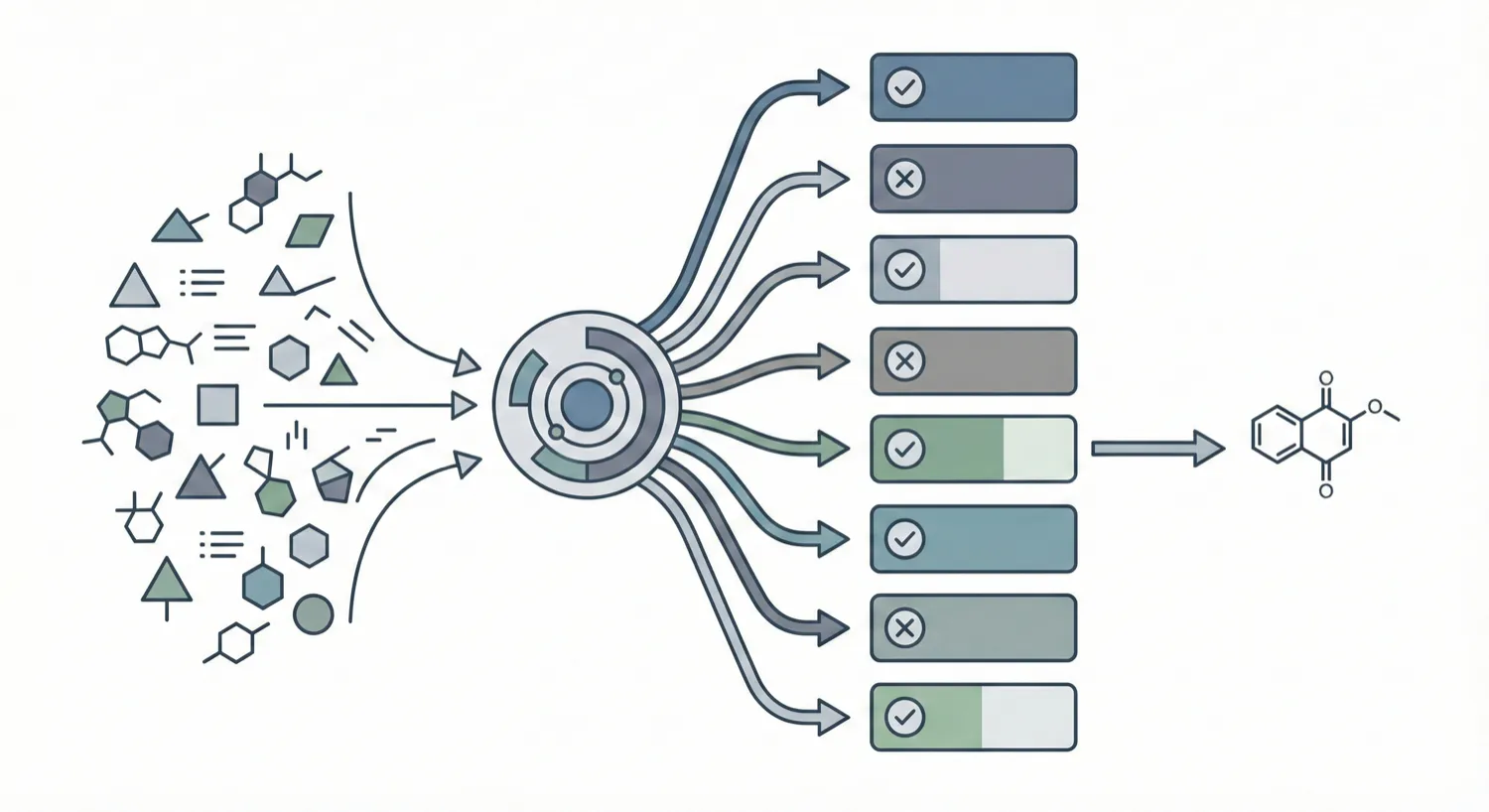
Comprehensive evaluation of 8 optical chemical structure recognition tools using a newly curated dataset of 2,702 patent images. Proposes ChemIC, a ResNet-50 classifier to route images to specialized tools based on content type, demonstrating that no single tool excels at all tasks.
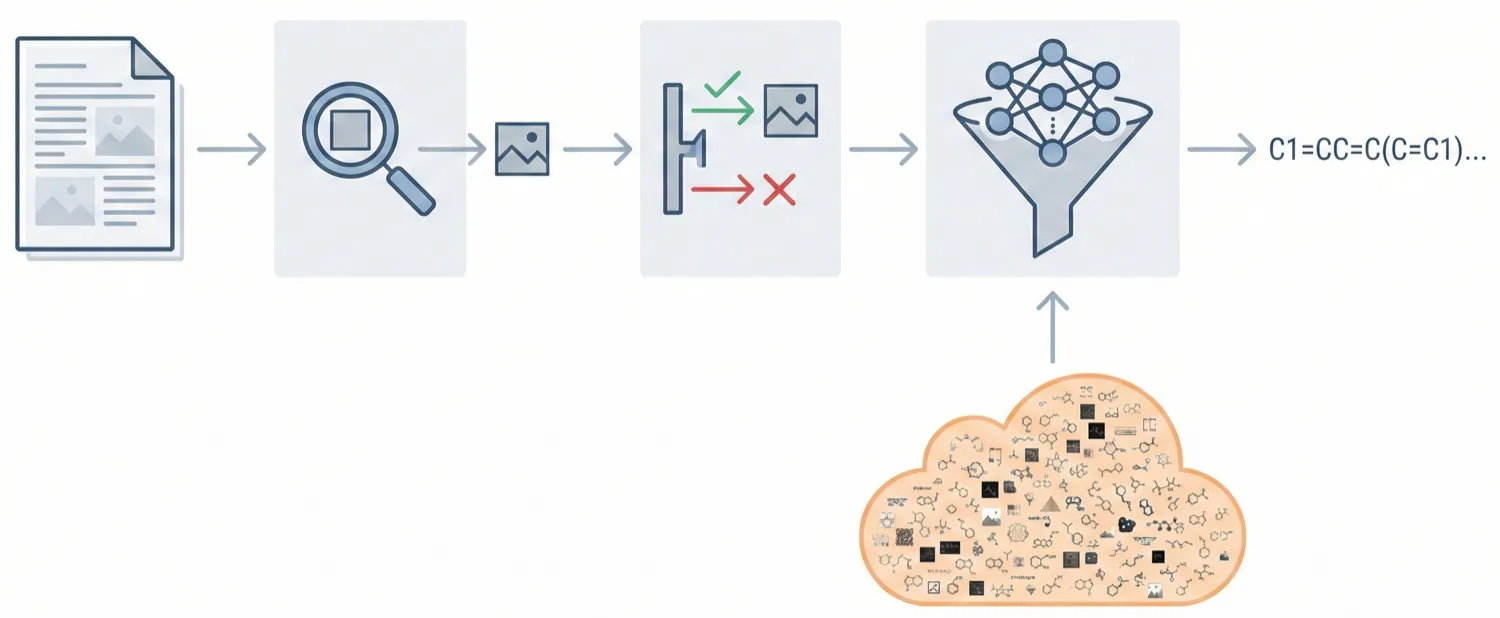
DECIMER.ai addresses the lack of open tools for Optical Chemical Structure Recognition (OCSR) by providing a comprehensive, deep-learning-based workflow. It features a novel data generation pipeline (RanDepict), a web application, and models for segmentation and recognition that rival or exceed proprietary solutions.
MolMiner replaces traditional rule-based vectorization with a deep learning object detection pipeline (YOLOv5) to extract chemical structures from PDFs. It outperforms open-source baselines on four benchmarks and introduces a new real-world dataset of 3,040 images.