Coscientist: Autonomous Chemistry with LLM Agents
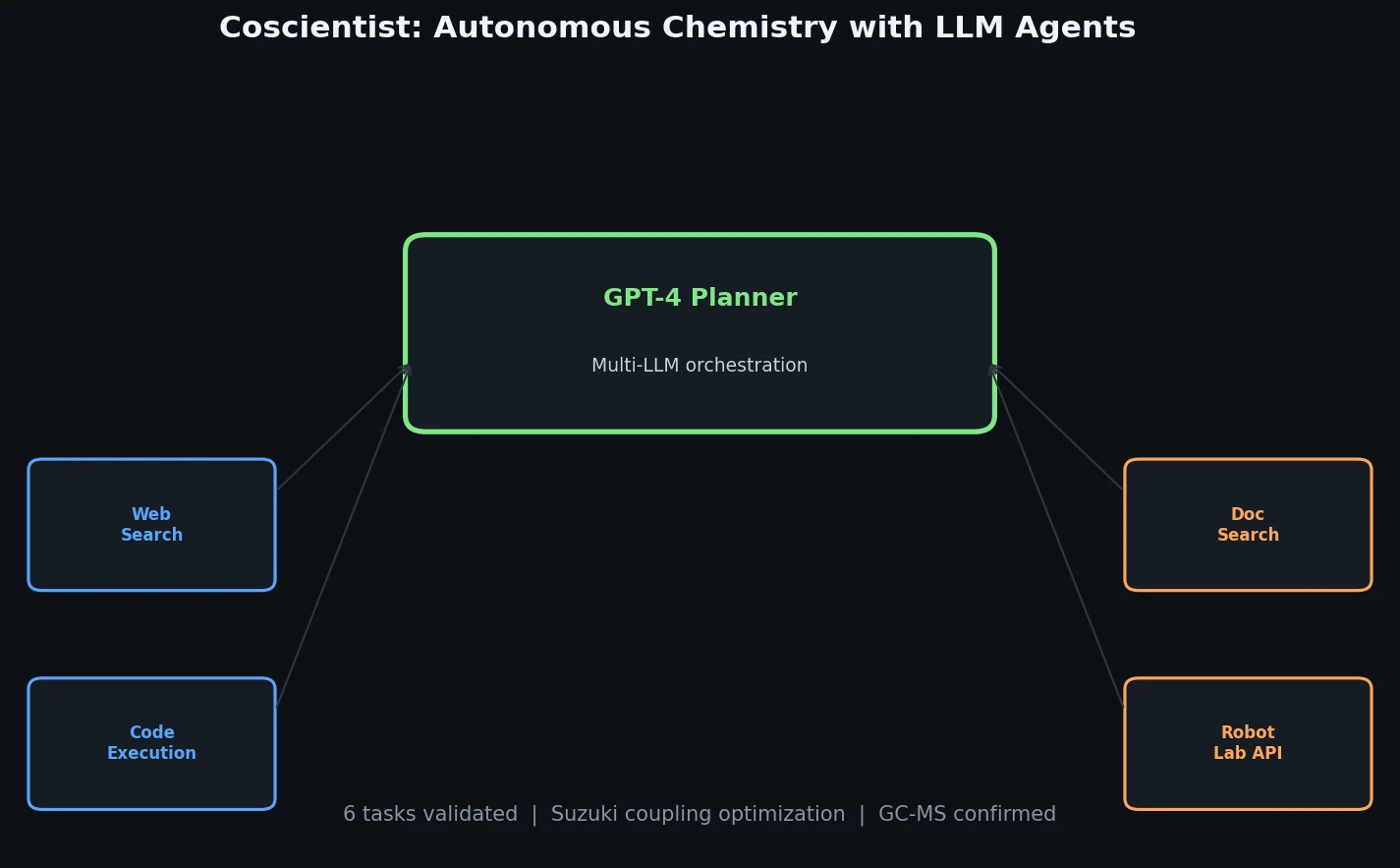
Introduces Coscientist, a GPT-4-driven AI system that autonomously designs and executes chemical experiments using web search, code execution, and robotic lab automation.
Introduces Coscientist, a GPT-4-driven AI system that autonomously designs and executes chemical experiments using web search, code execution, and robotic lab automation.
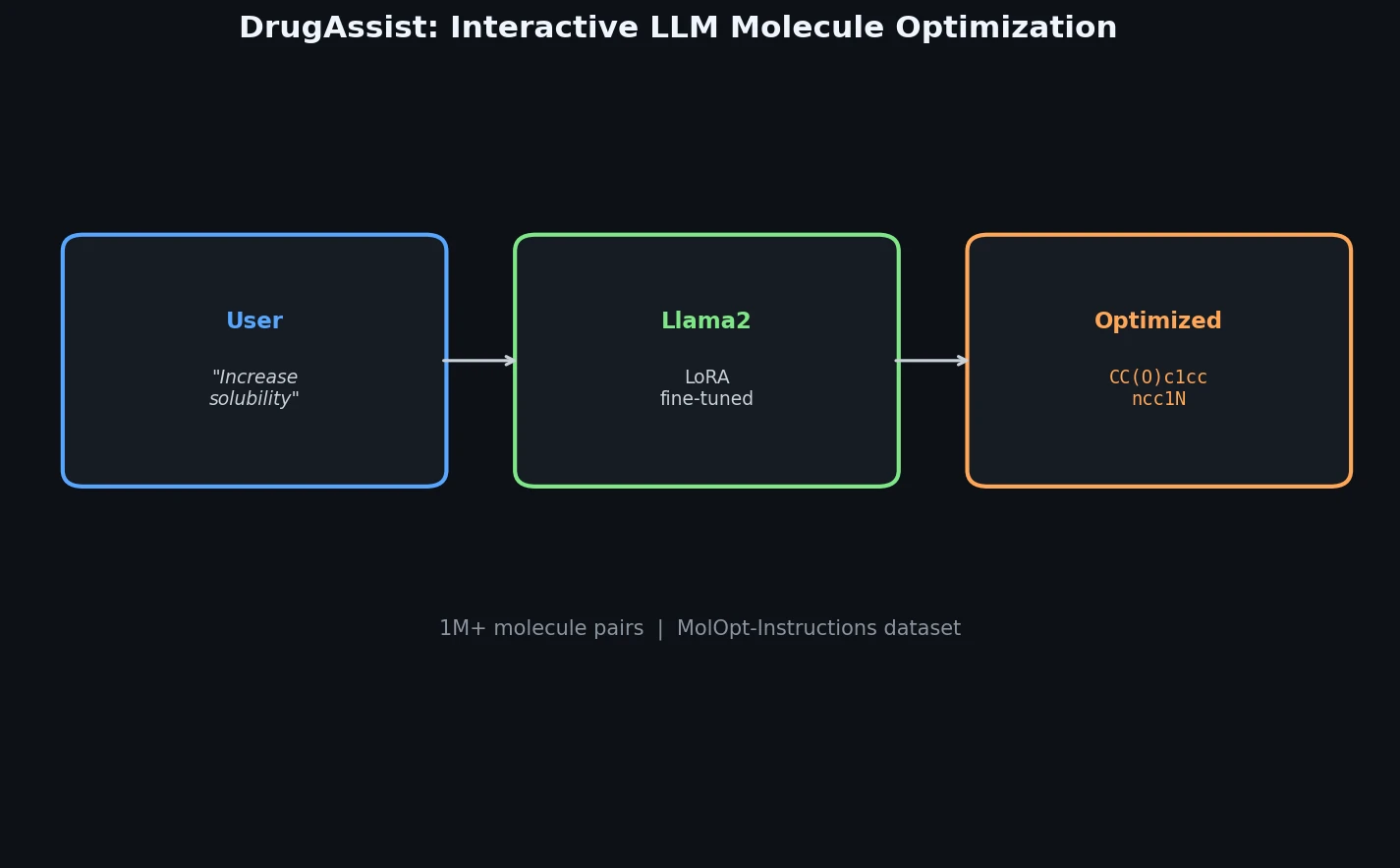
DrugAssist fine-tunes Llama2-7B-Chat on over one million molecule pairs for interactive, dialogue-based molecule optimization across six molecular properties.
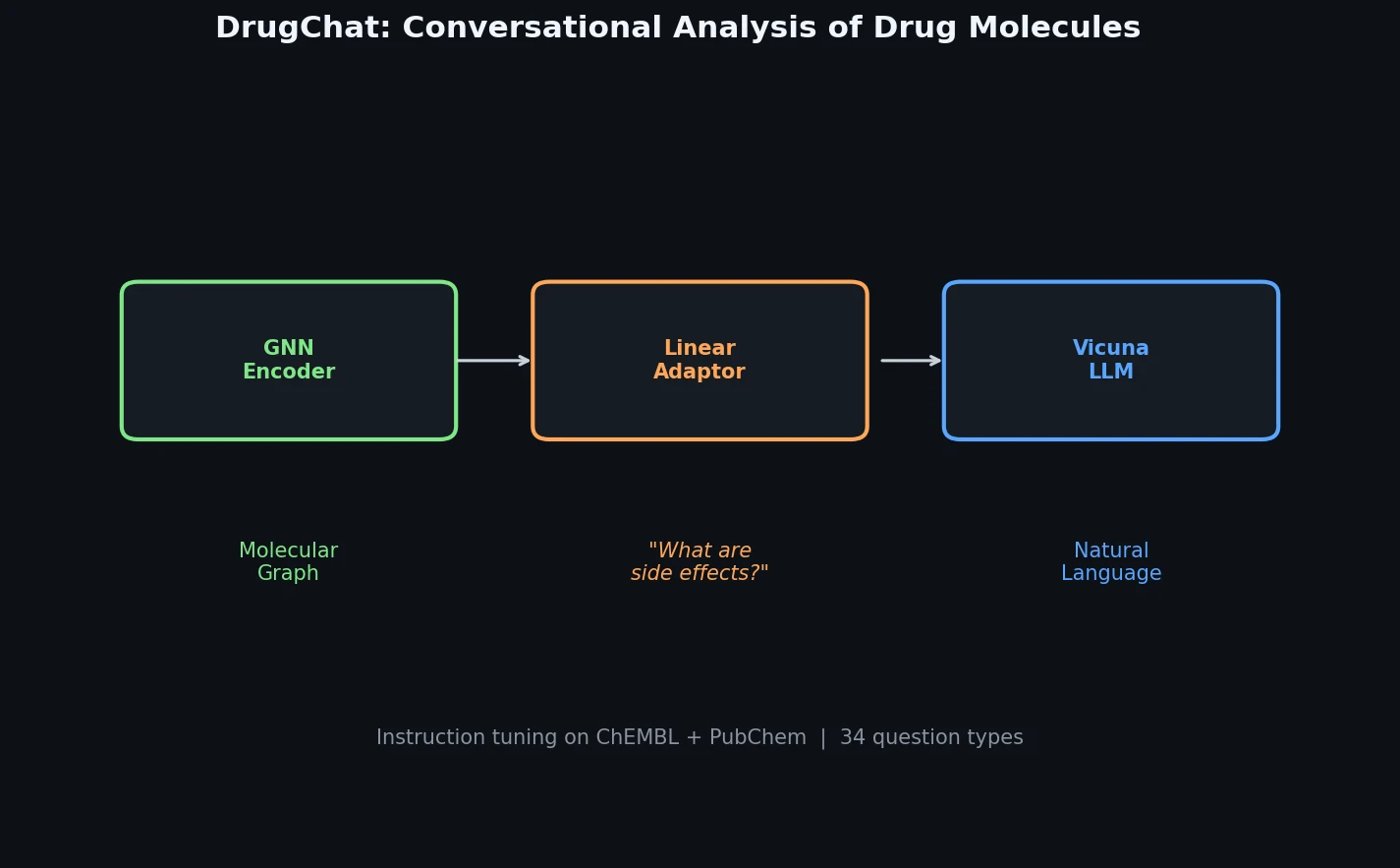
DrugChat is a prototype system that bridges molecular graph neural networks with large language models for interactive, multi-turn question answering about drug compounds. It trains only a lightweight linear adaptor between a frozen GNN encoder and Vicuna-13B using 143K curated QA pairs from ChEMBL and PubChem.
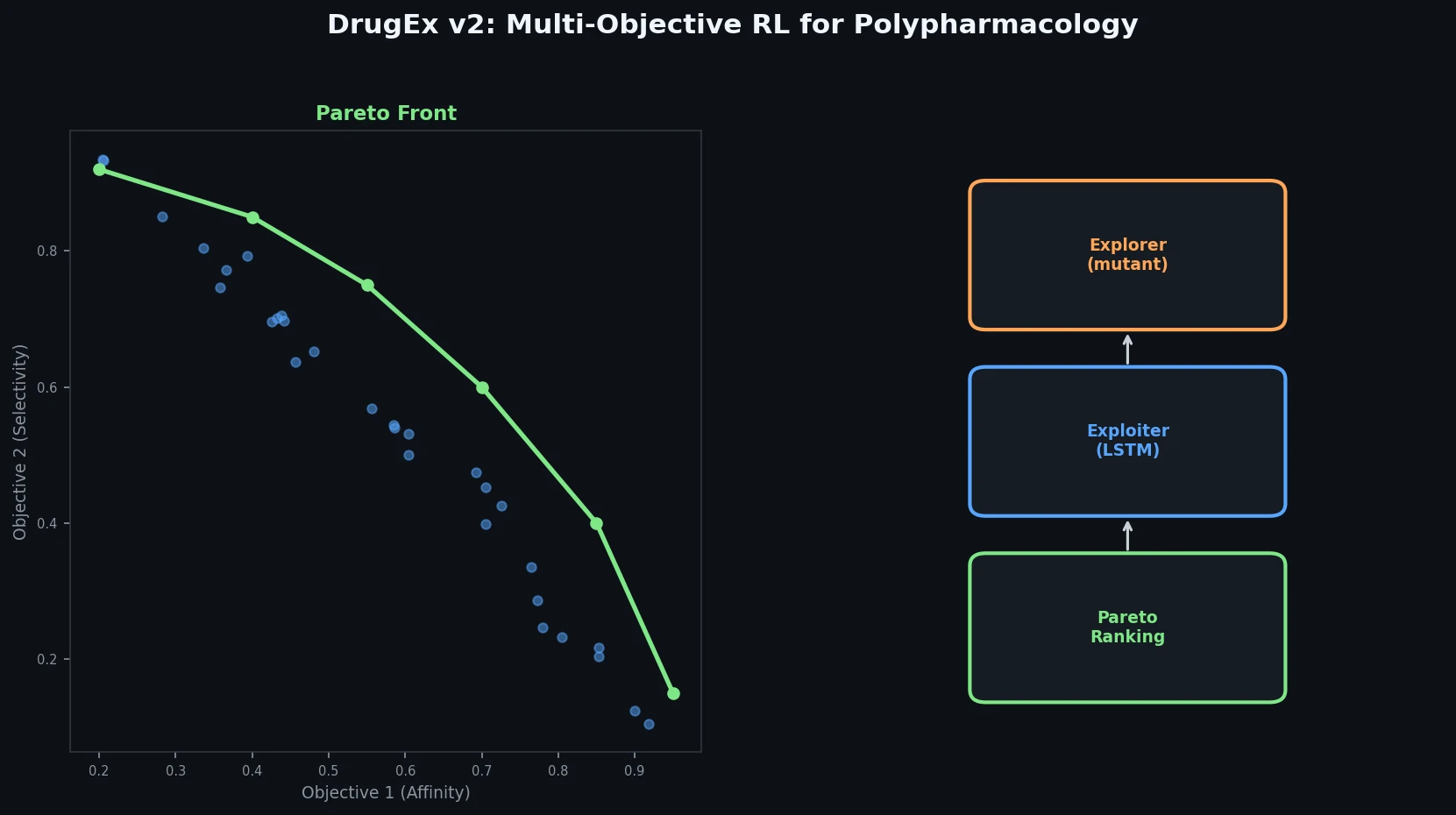
DrugEx v2 introduces Pareto-based multi-objective optimization and evolutionary exploration strategies into an RNN reinforcement learning framework for de novo drug design toward multiple protein targets.
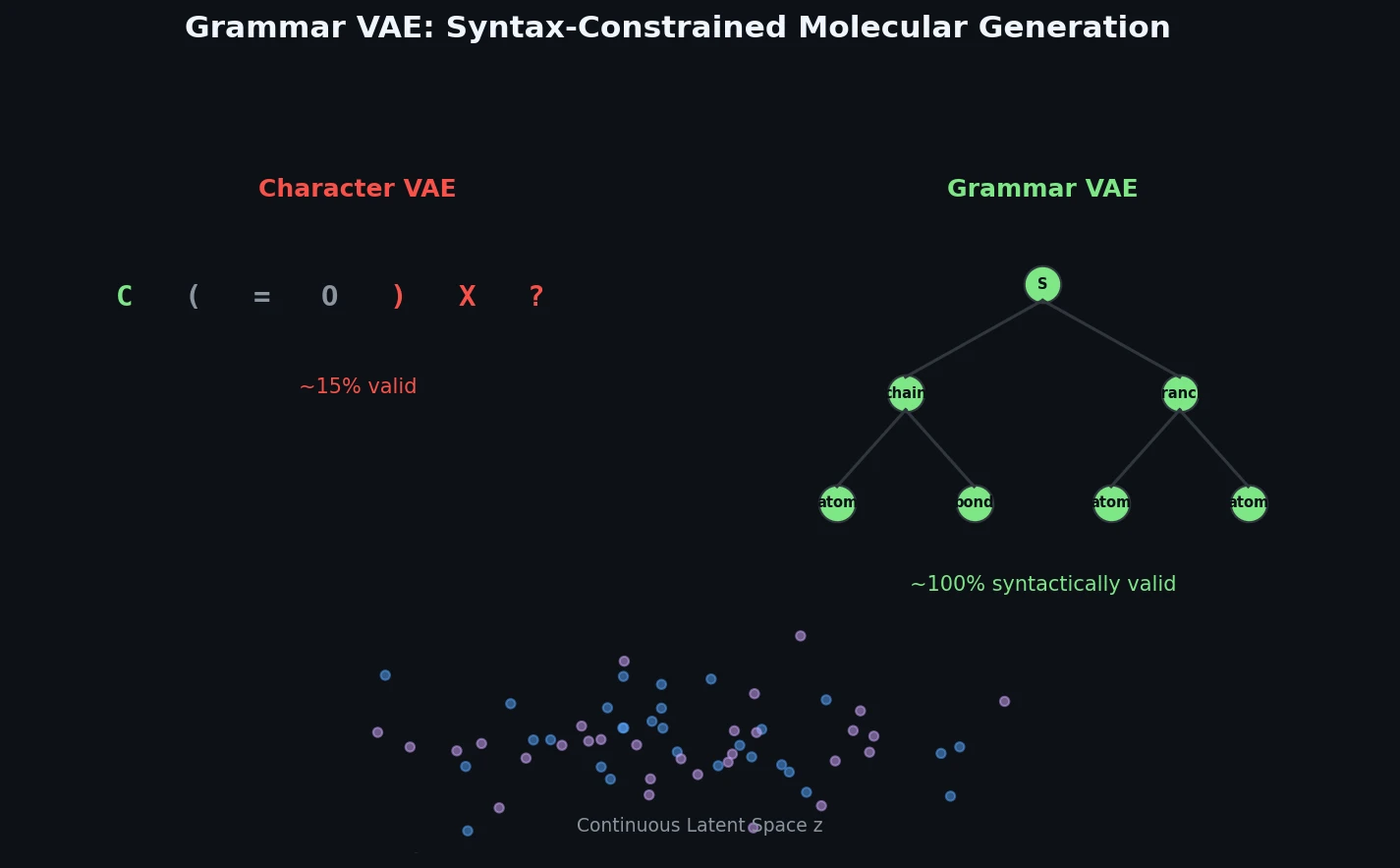
The Grammar VAE replaces character-level decoding with context-free grammar production rules, using a stack-based masking mechanism to guarantee that all generated SMILES strings are syntactically valid. Applied to molecular optimization and symbolic regression, it learns smoother latent spaces and finds better molecules than character-level baselines.
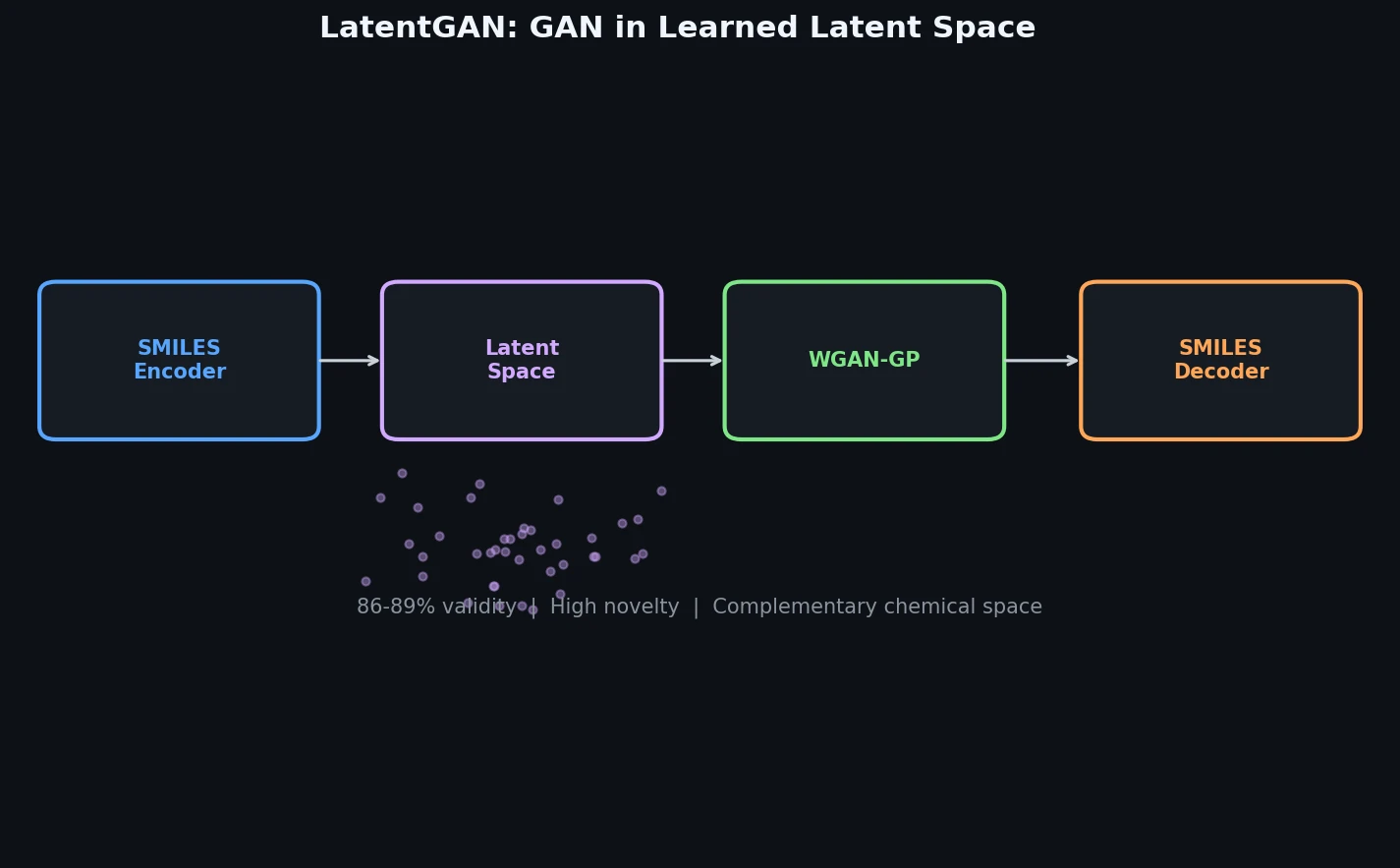
LatentGAN decouples molecular generation from SMILES syntax by training a Wasserstein GAN on latent vectors from a pretrained heteroencoder, enabling de novo design of drug-like and target-biased compounds.
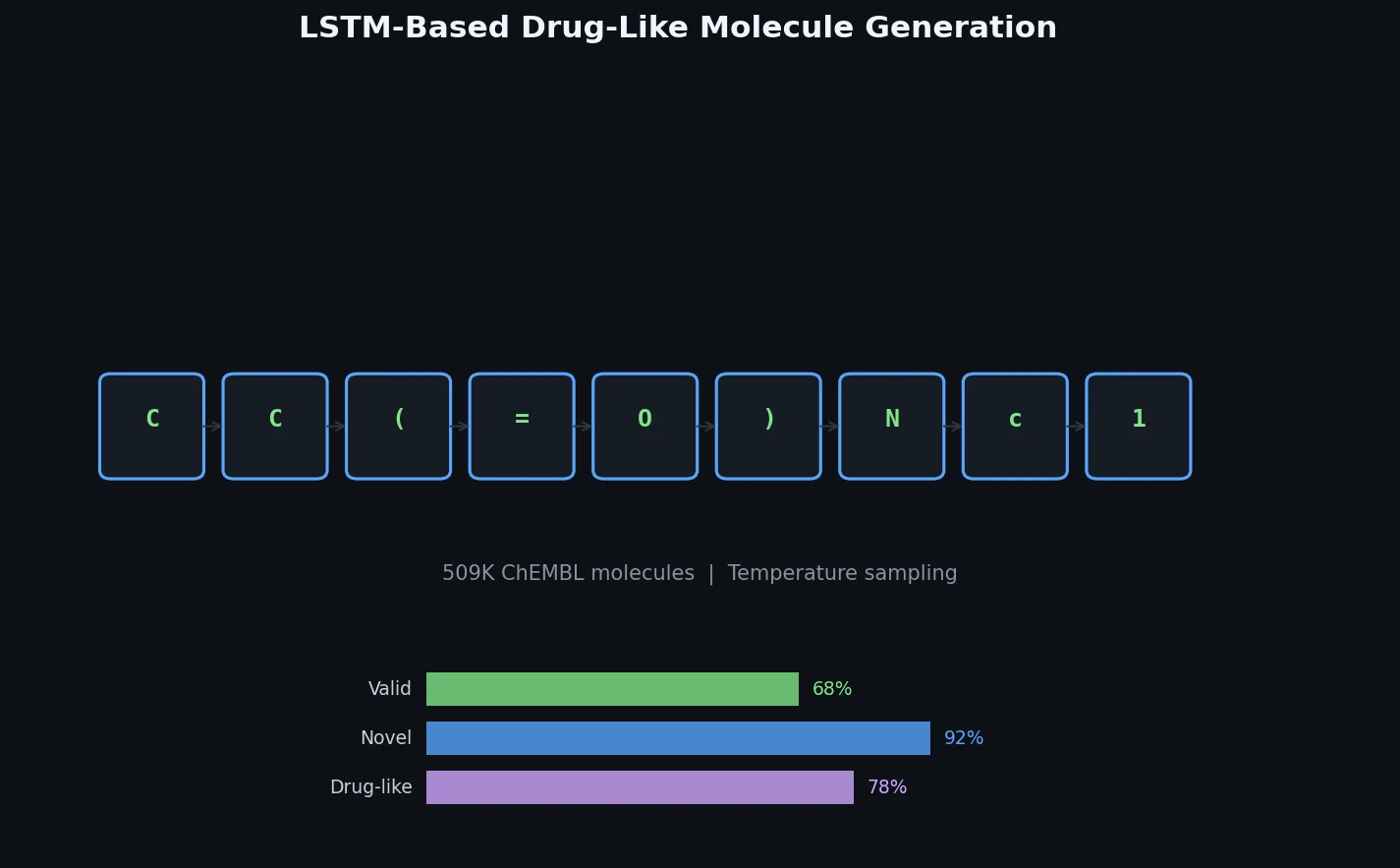
Ertl et al. train a character-level LSTM on 509K bioactive ChEMBL SMILES and generate one million novel, diverse molecules whose physicochemical properties, substructure features, and predicted bioactivity closely match the training distribution.
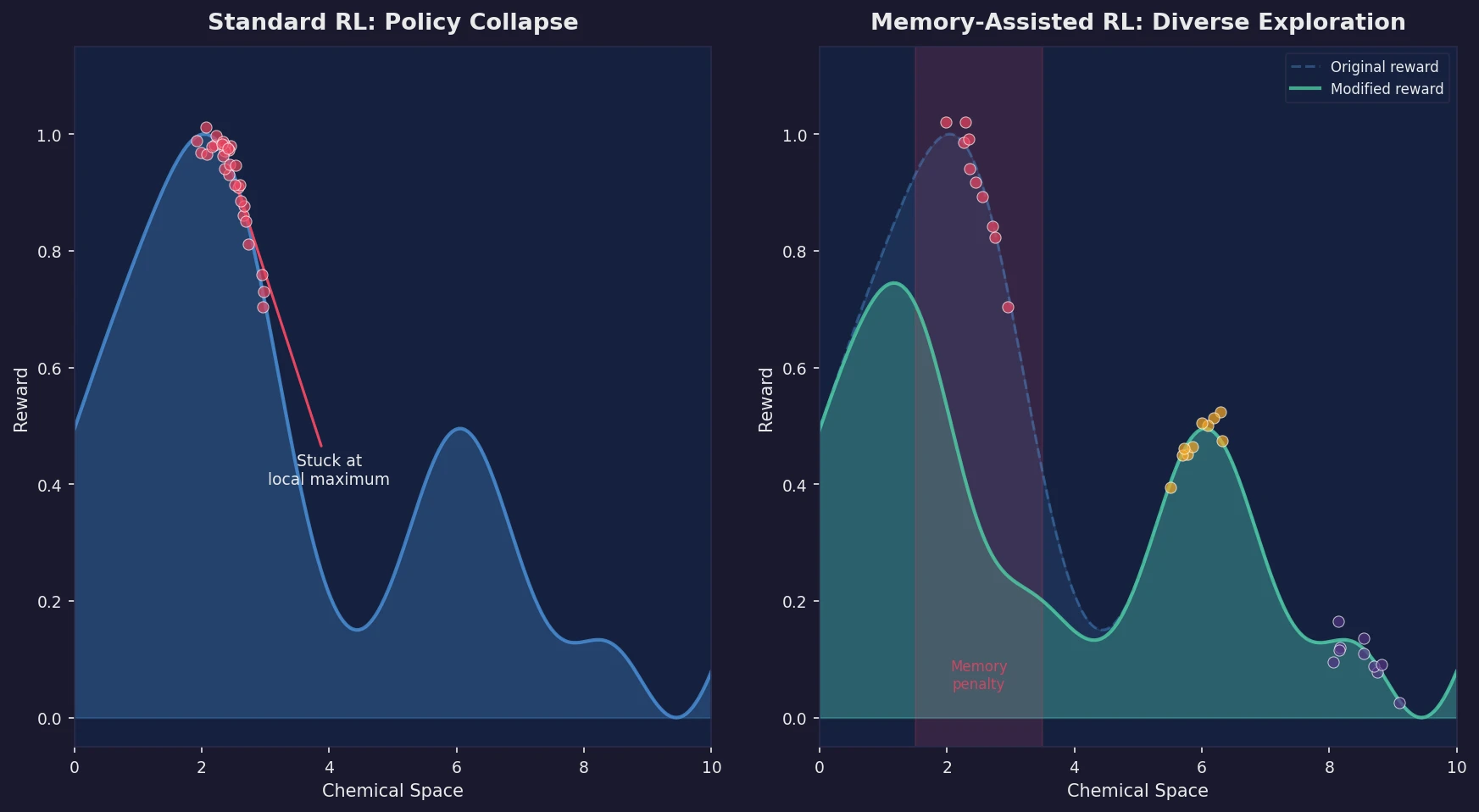
Introduces a memory unit that modifies the RL reward function to penalize previously explored chemical scaffolds, substantially increasing the diversity of generated molecules while maintaining relevance to known active ligands.
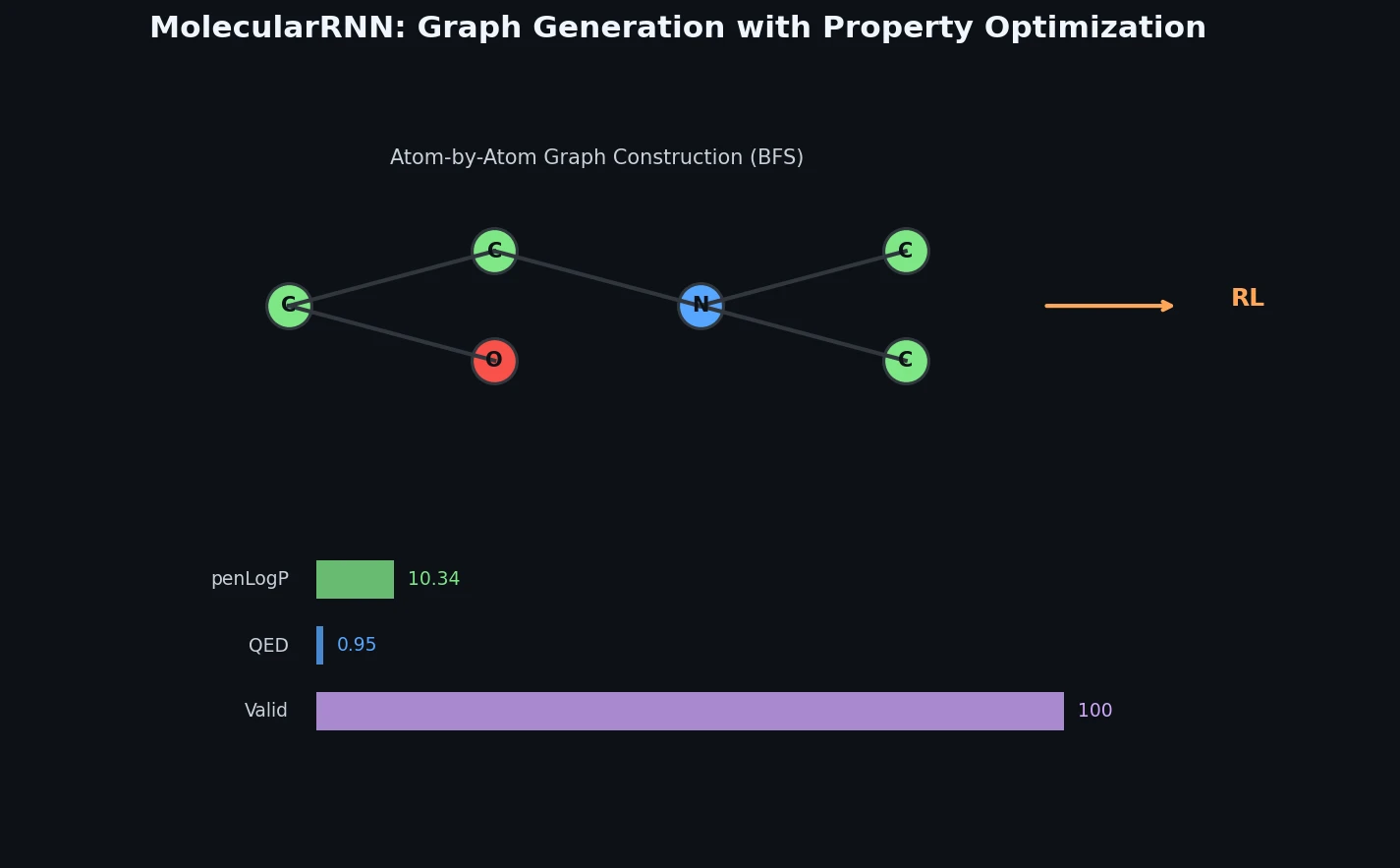
Proposes MolecularRNN, a graph recurrent model that generates molecular graphs atom-by-atom with 100% validity via valency-based rejection sampling, then shifts property distributions using policy gradient reinforcement learning.
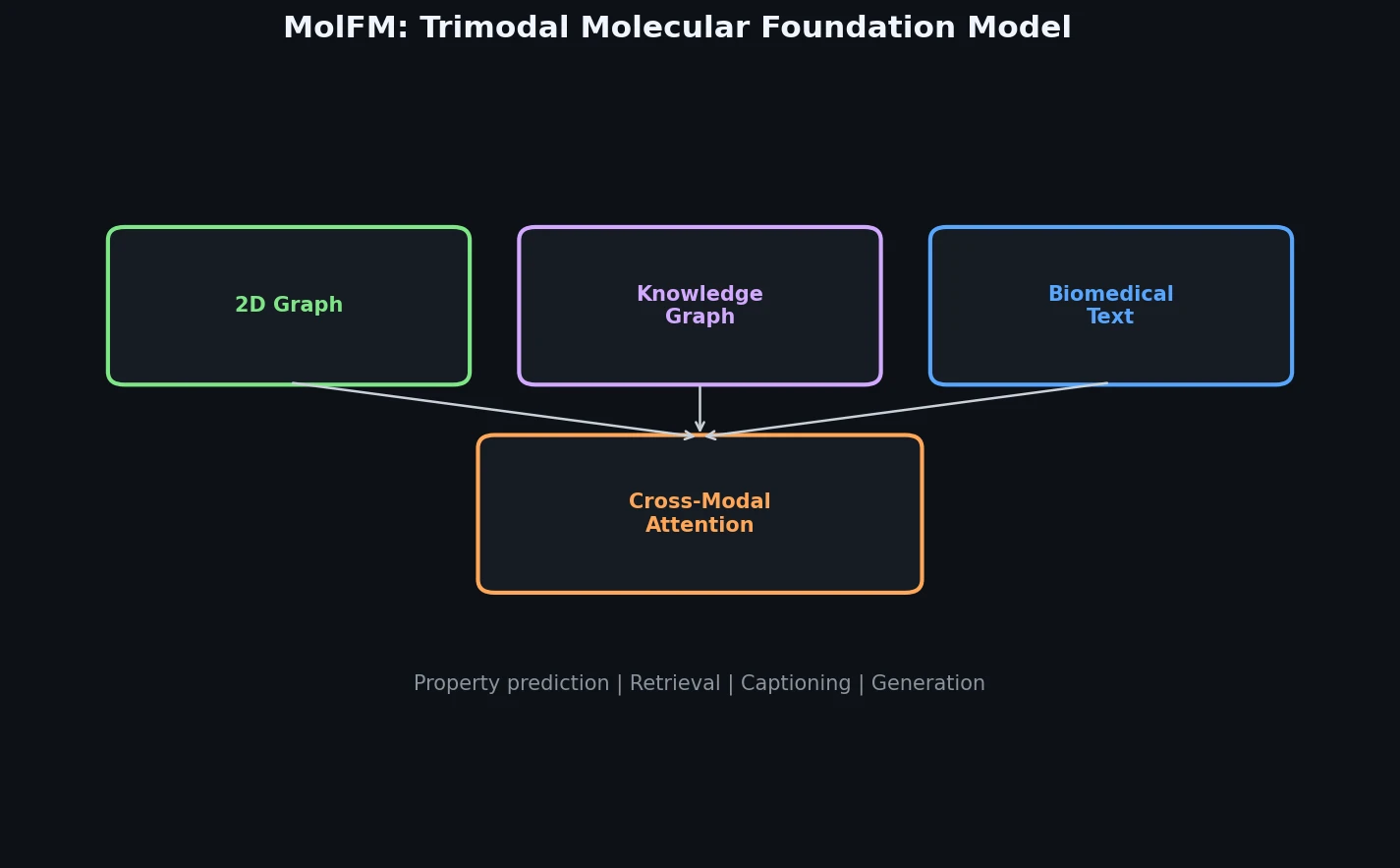
MolFM pre-trains a multimodal encoder that fuses 2D molecular graphs, biomedical text, and knowledge graph entities through fine-grained cross-modal attention, achieving strong gains on cross-modal retrieval, molecule captioning, text-based generation, and property prediction.
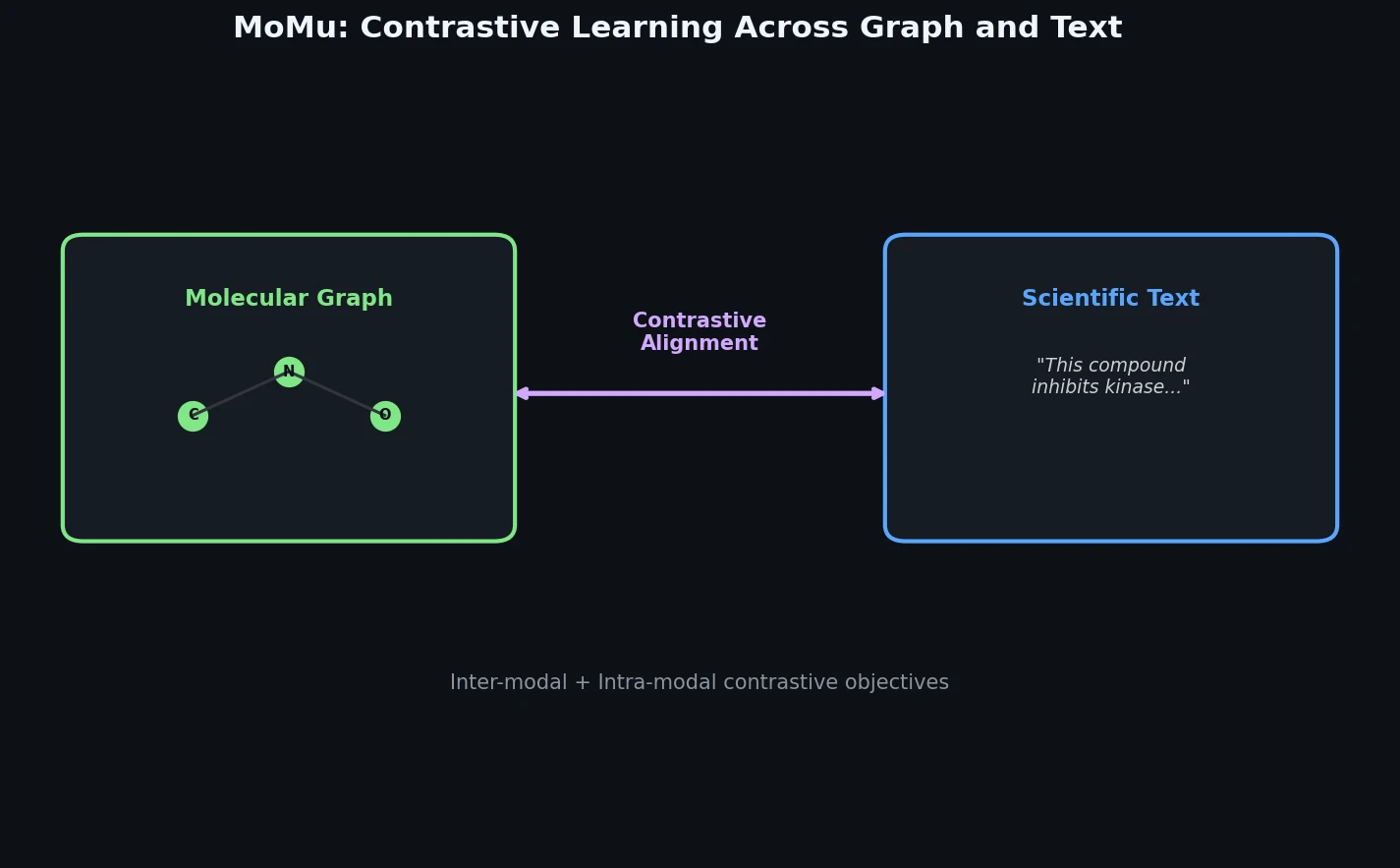
MoMu pre-trains dual graph and text encoders on 15K molecule graph-text pairs using contrastive learning, enabling cross-modal retrieval, molecule captioning, zero-shot text-to-graph generation, and improved molecular property prediction.
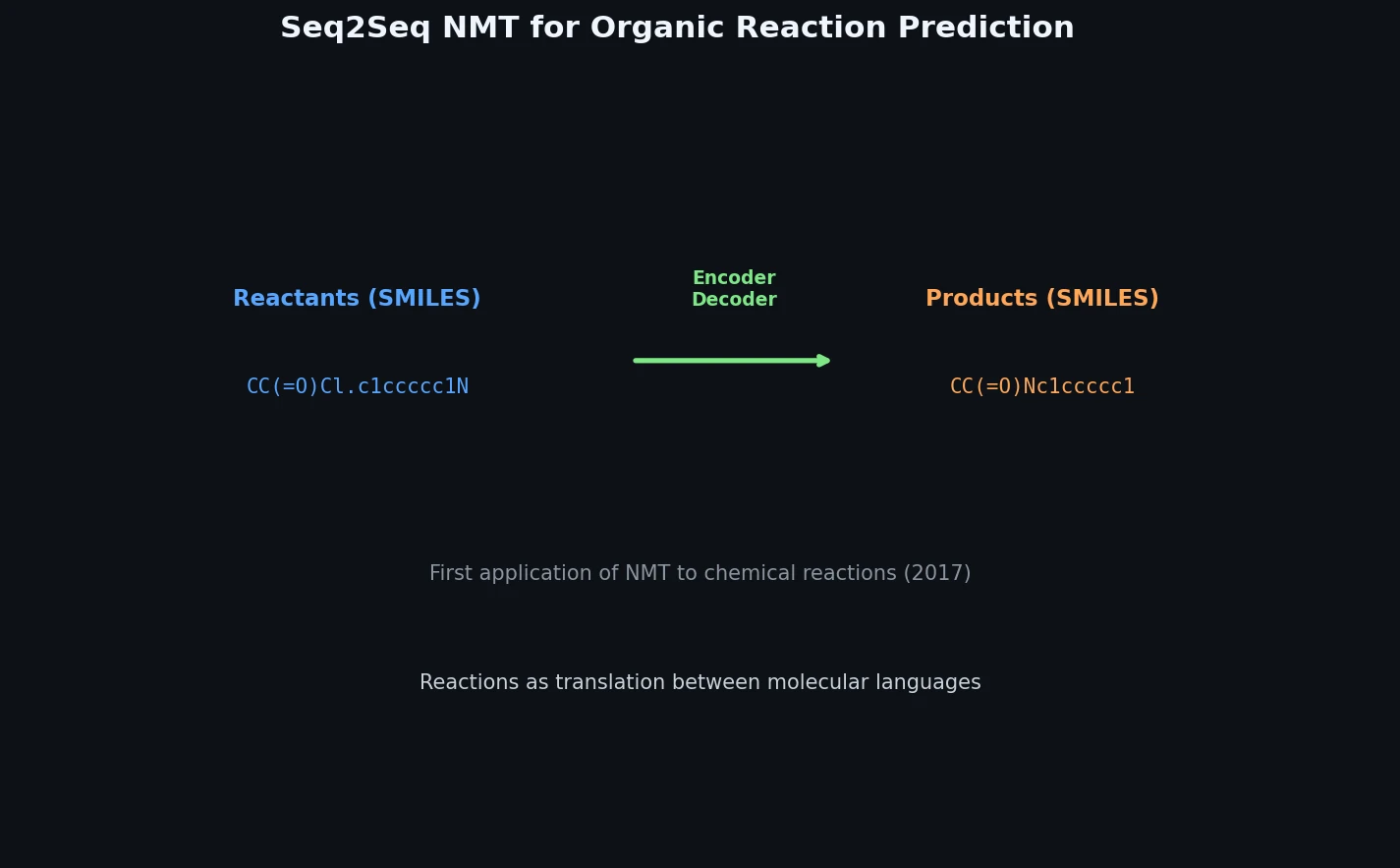
This 2016 paper first proposed treating organic reaction prediction as a neural machine translation problem, using a GRU-based sequence-to-sequence model with attention to translate reactant SMILES strings into product SMILES strings.