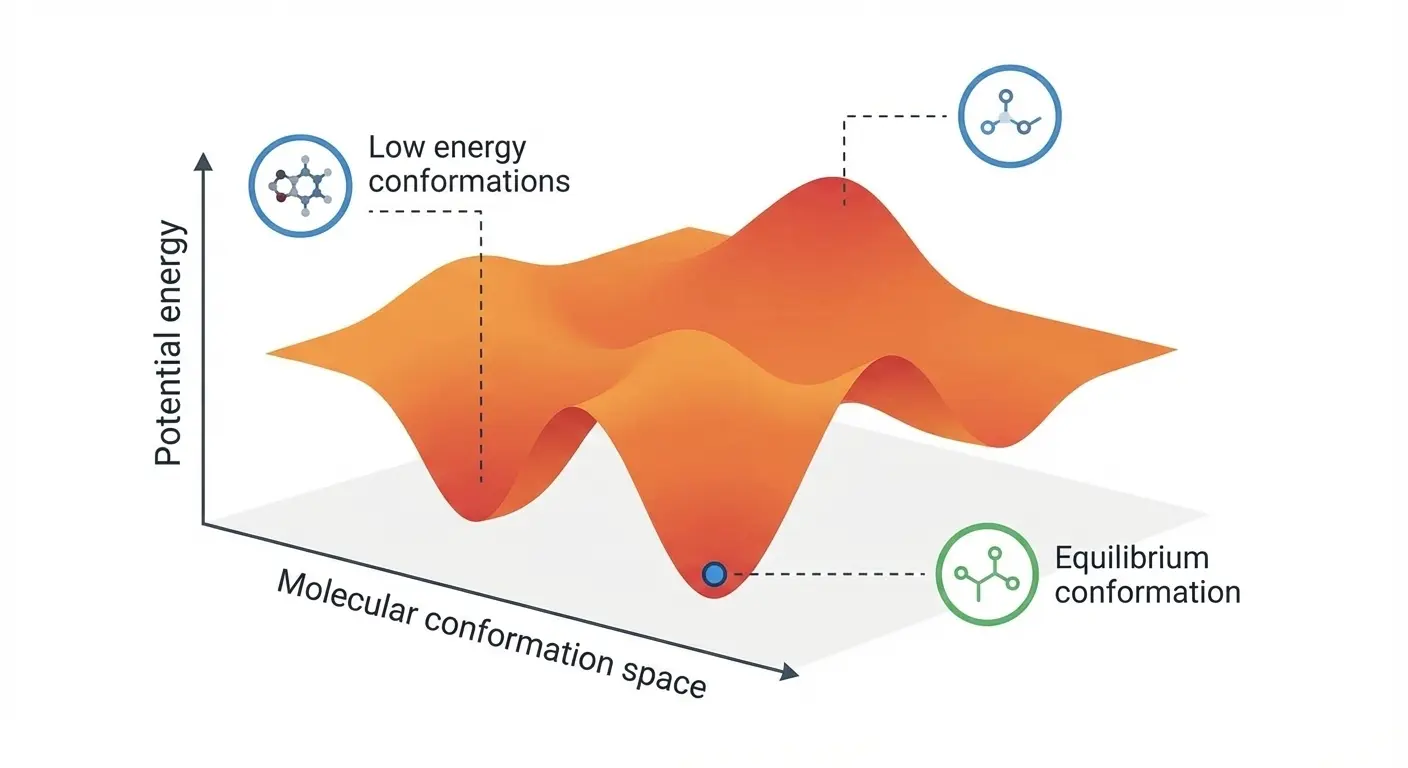
DenoiseVAE: Adaptive Noise for Molecular Pre-training
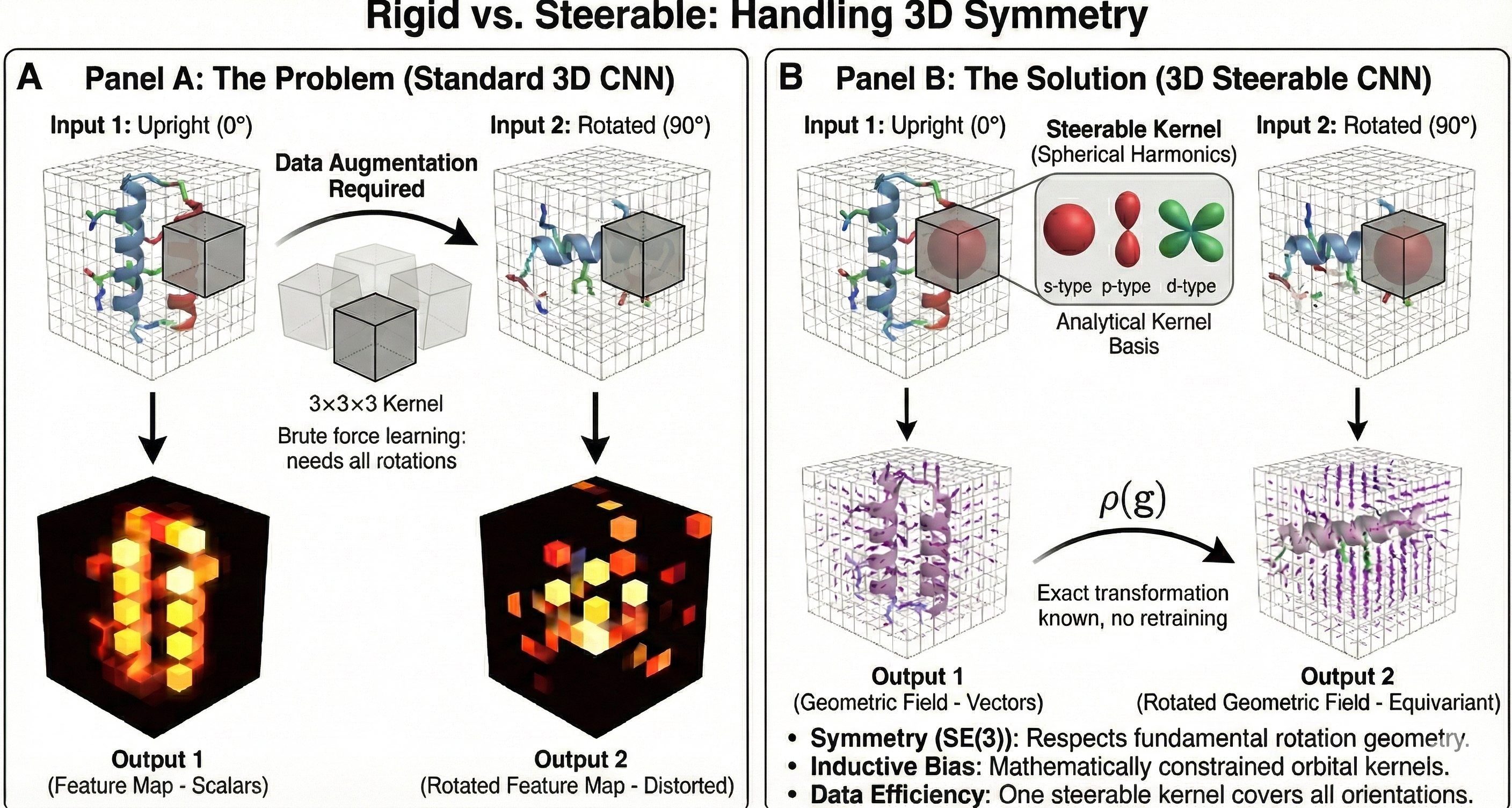
ICLR 2025 paper introducing DenoiseVAE, which learns adaptive, atom-specific noise distributions through a VAE framework to improve denoising-based pre-training for molecular force field prediction, outperforming fixed Gaussian noise approaches on quantum chemistry benchmarks.