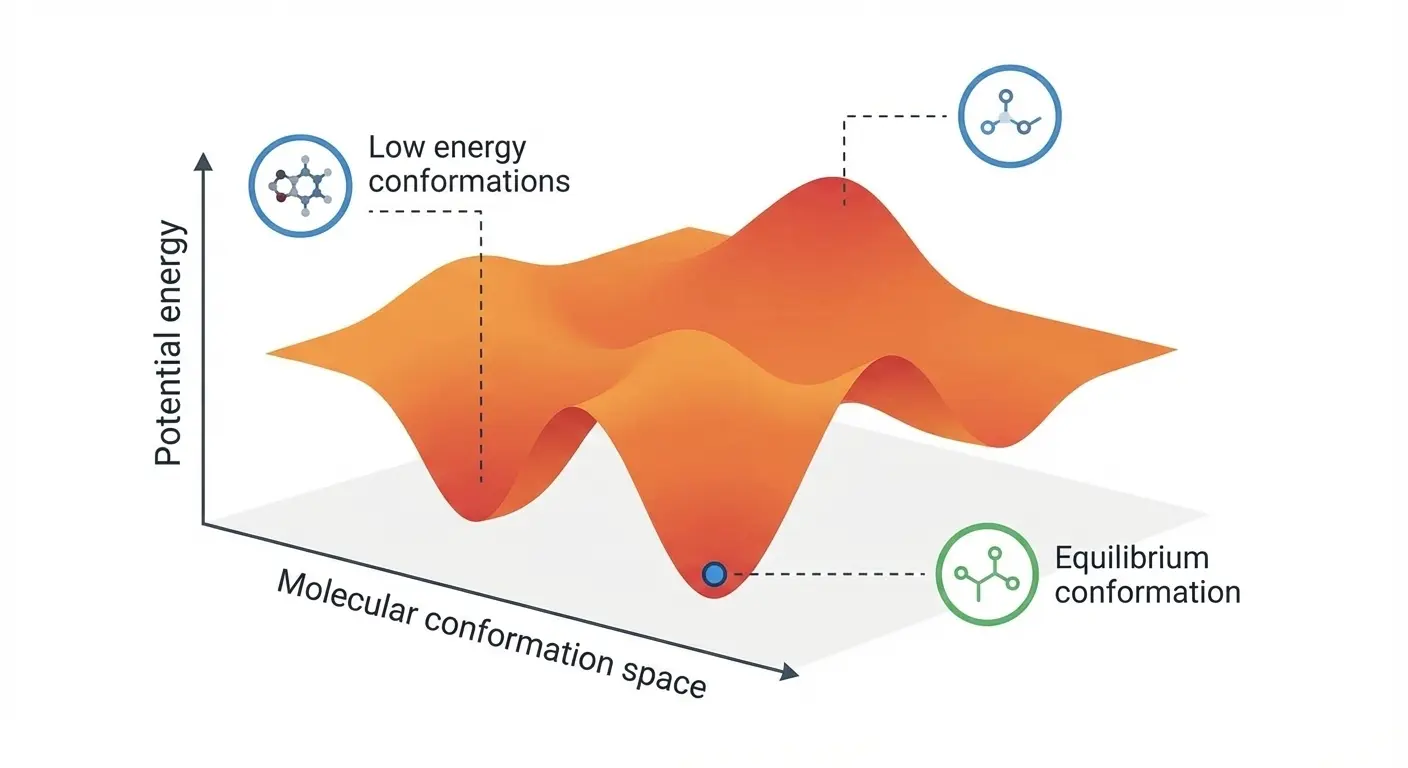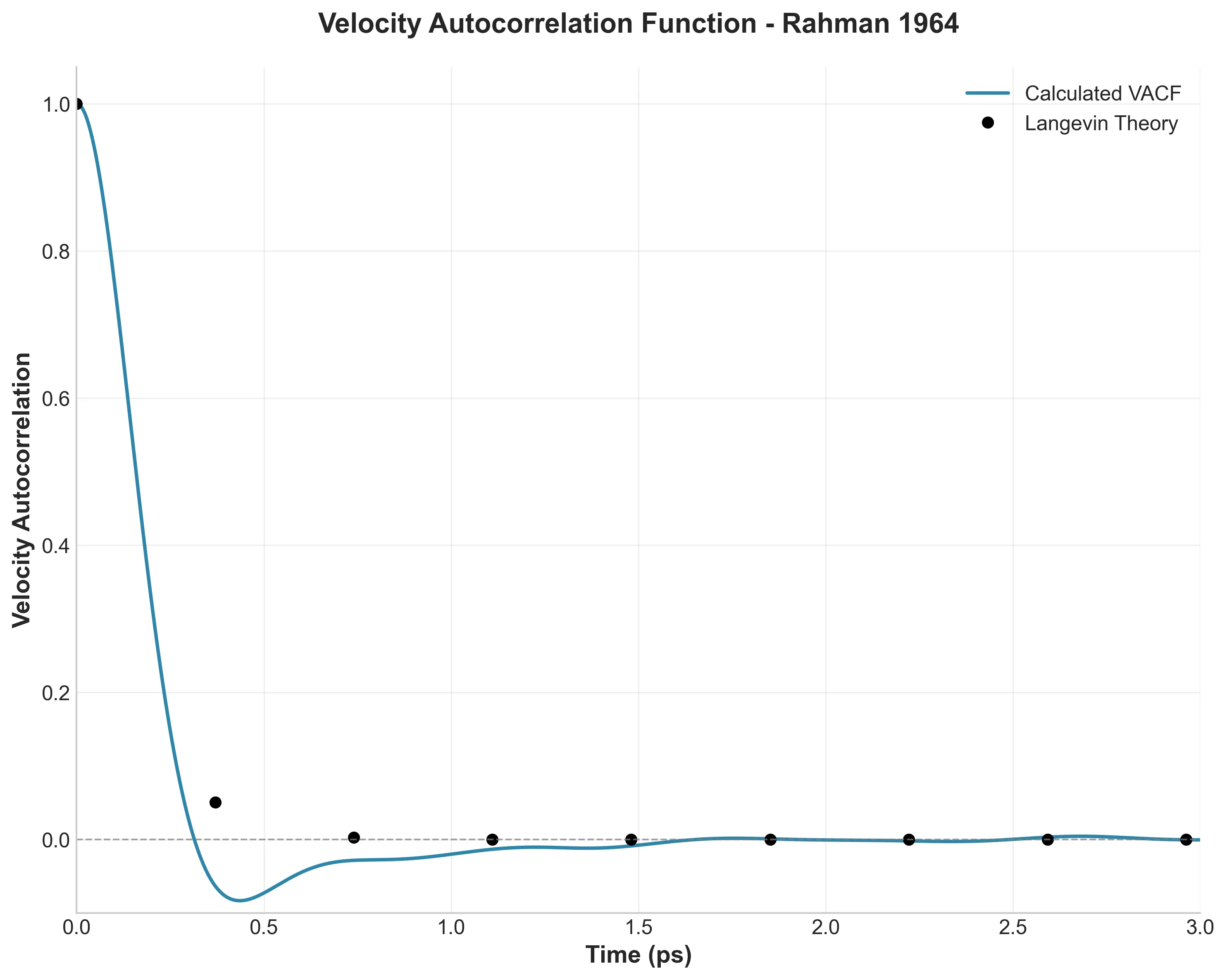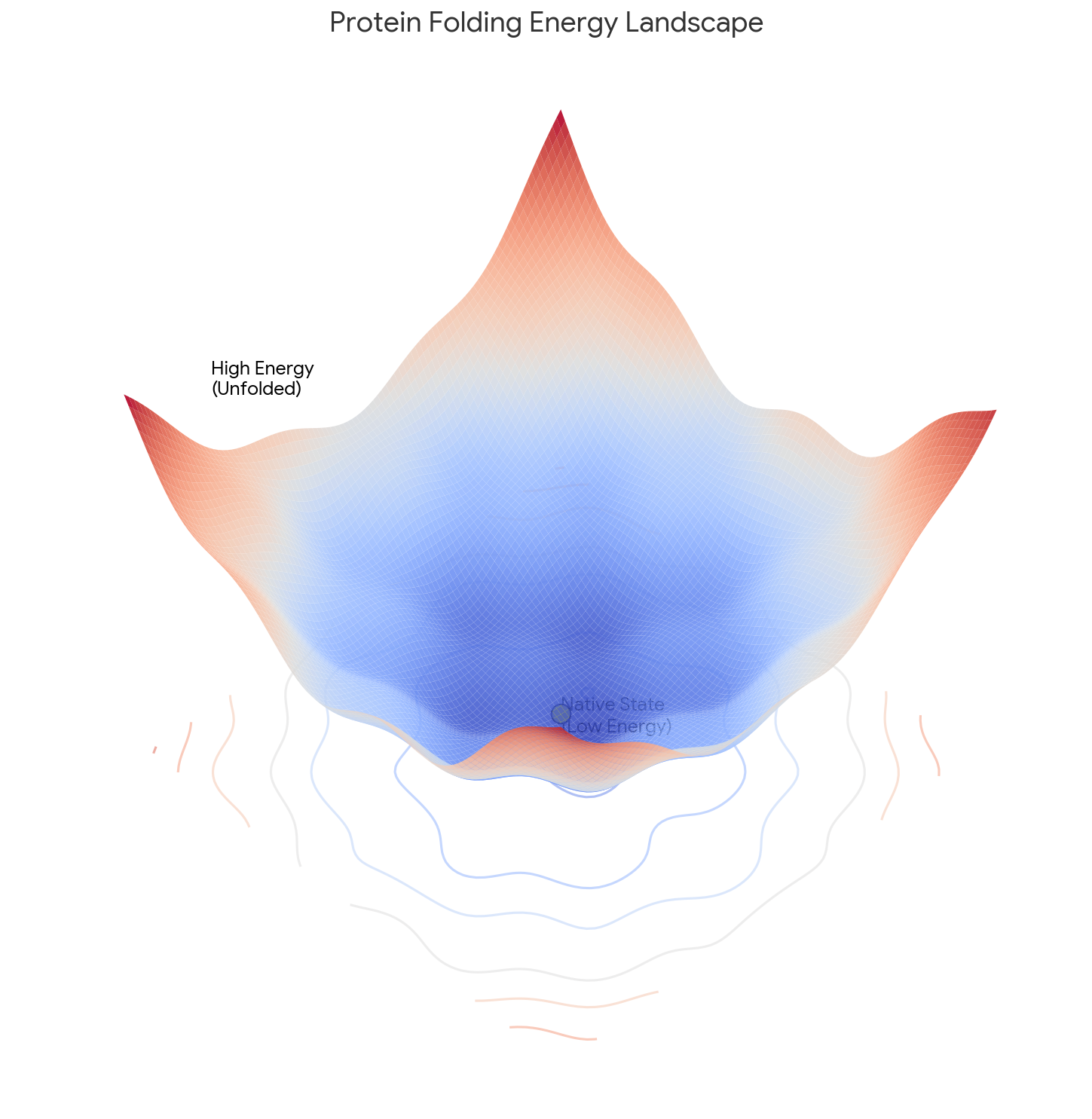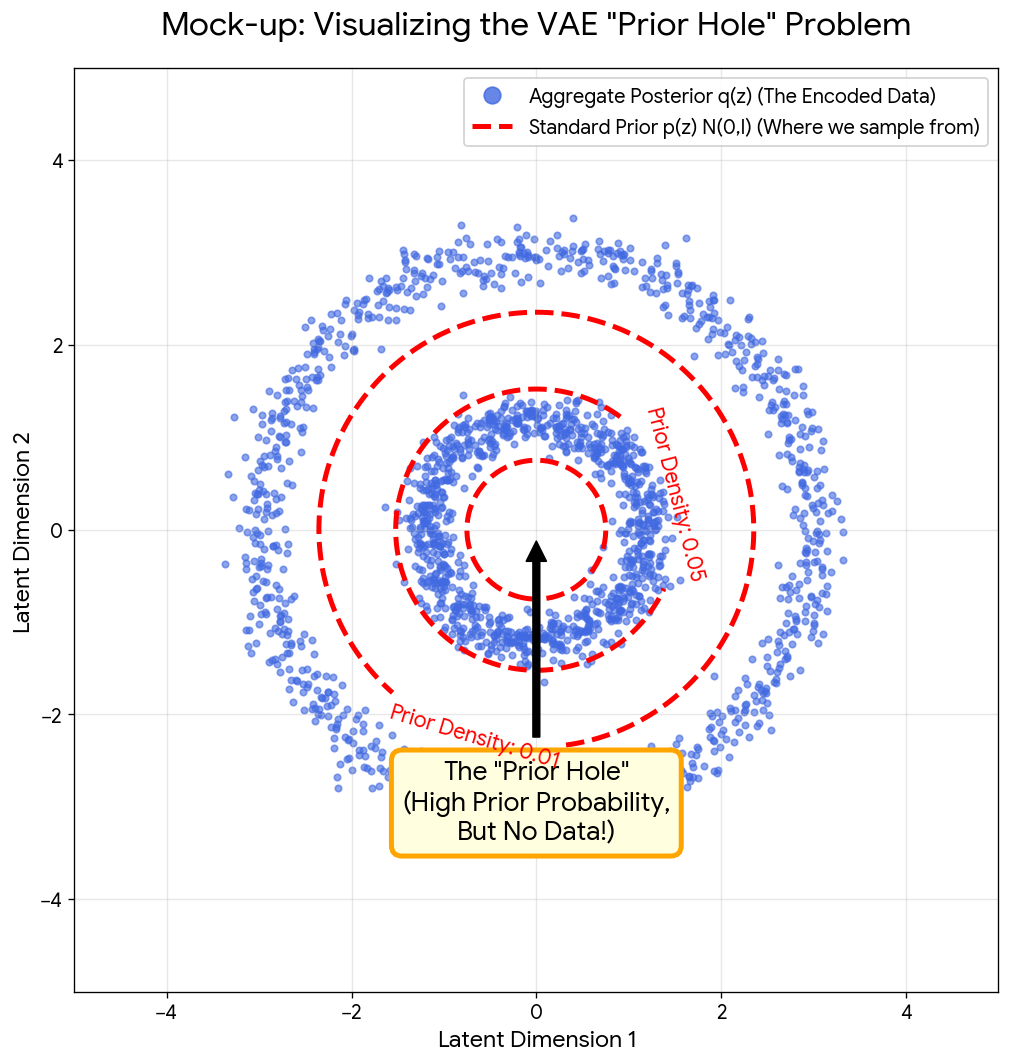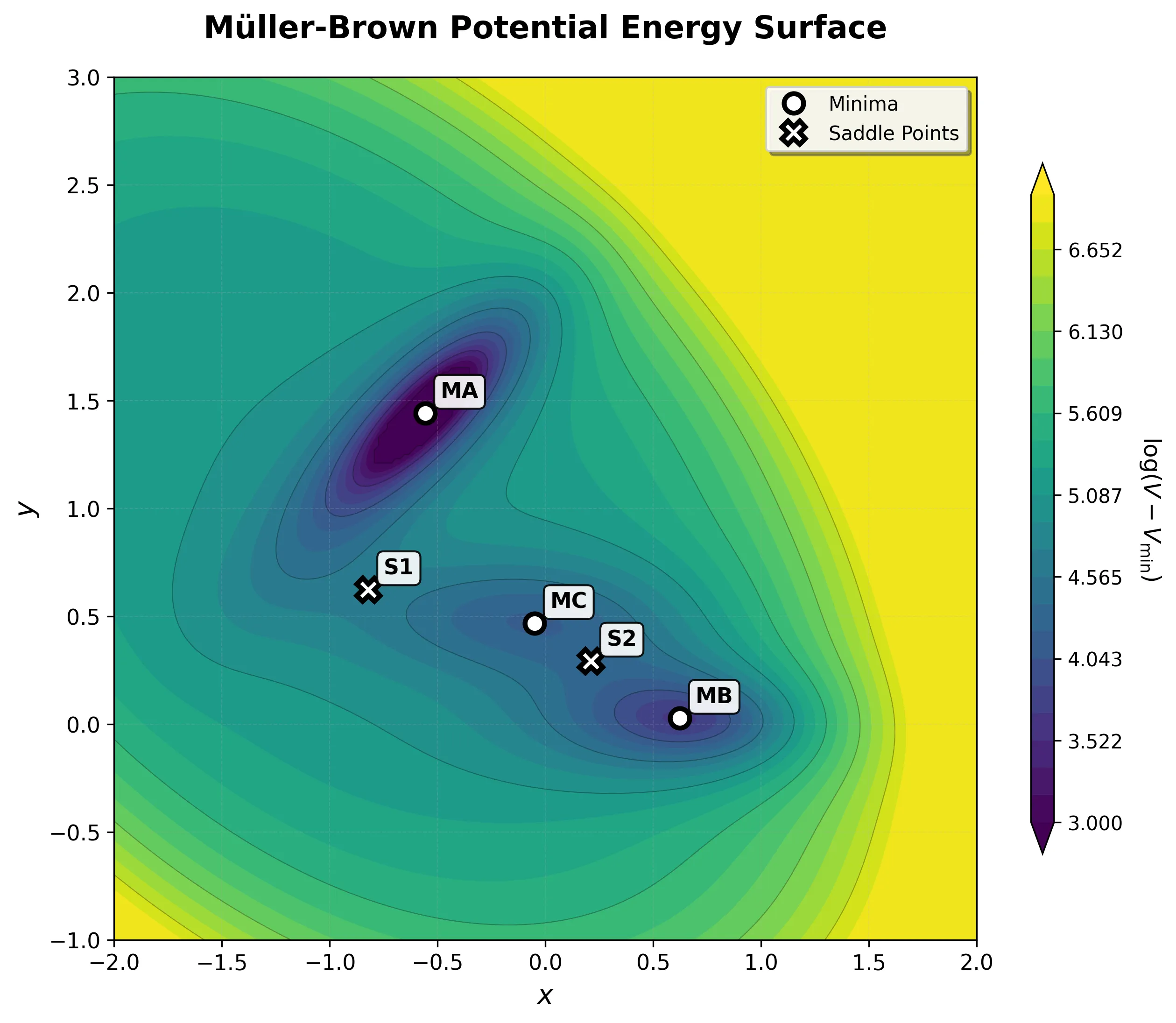
Implementing the Müller-Brown Potential in PyTorch
Step-by-step implementation of the classic Müller-Brown potential in PyTorch, with performance comparisons between analytical and automatic differentiation approaches for molecular dynamics and machine learning applications.