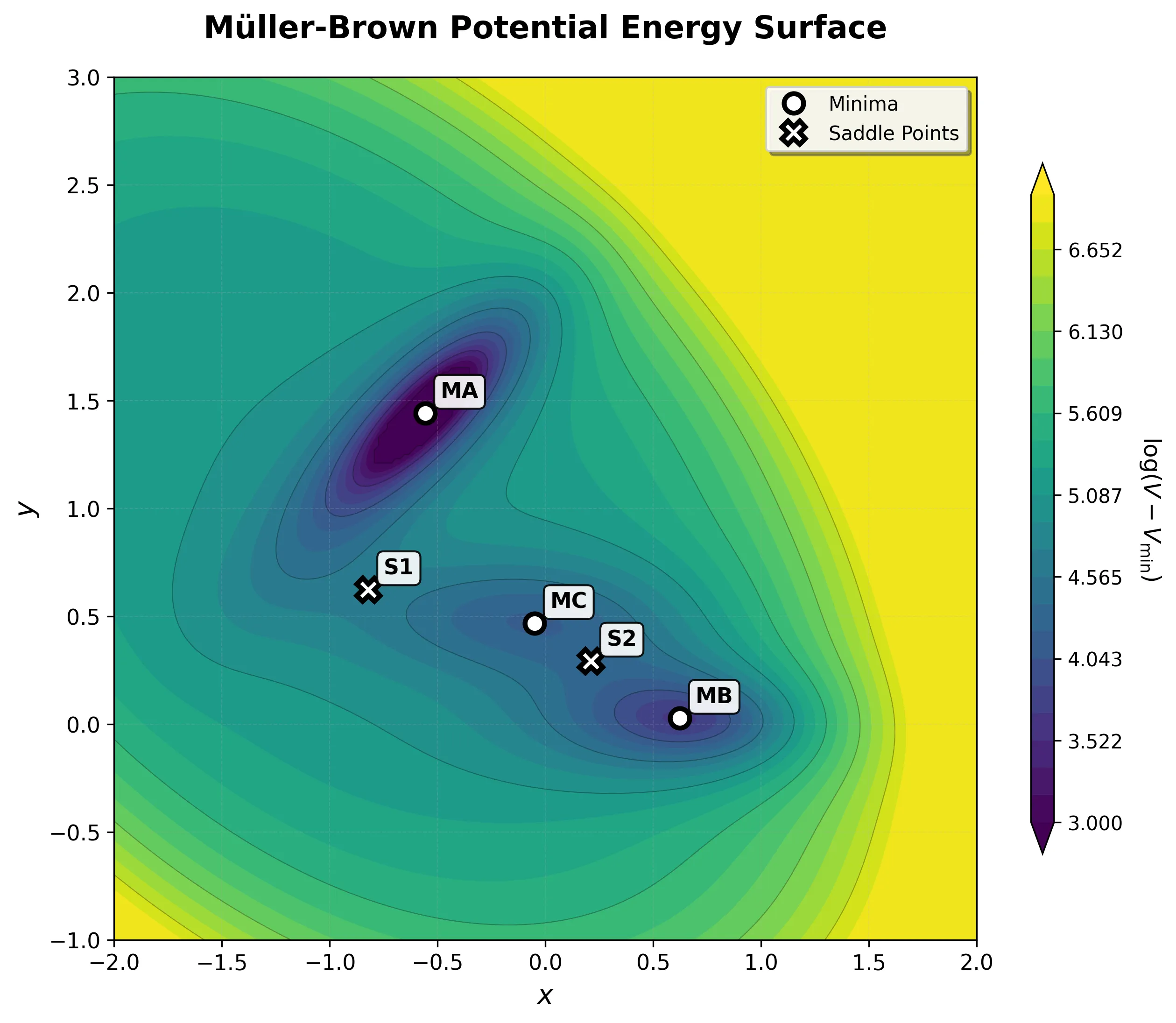
Müller-Brown Potential: A PyTorch ML Testbed
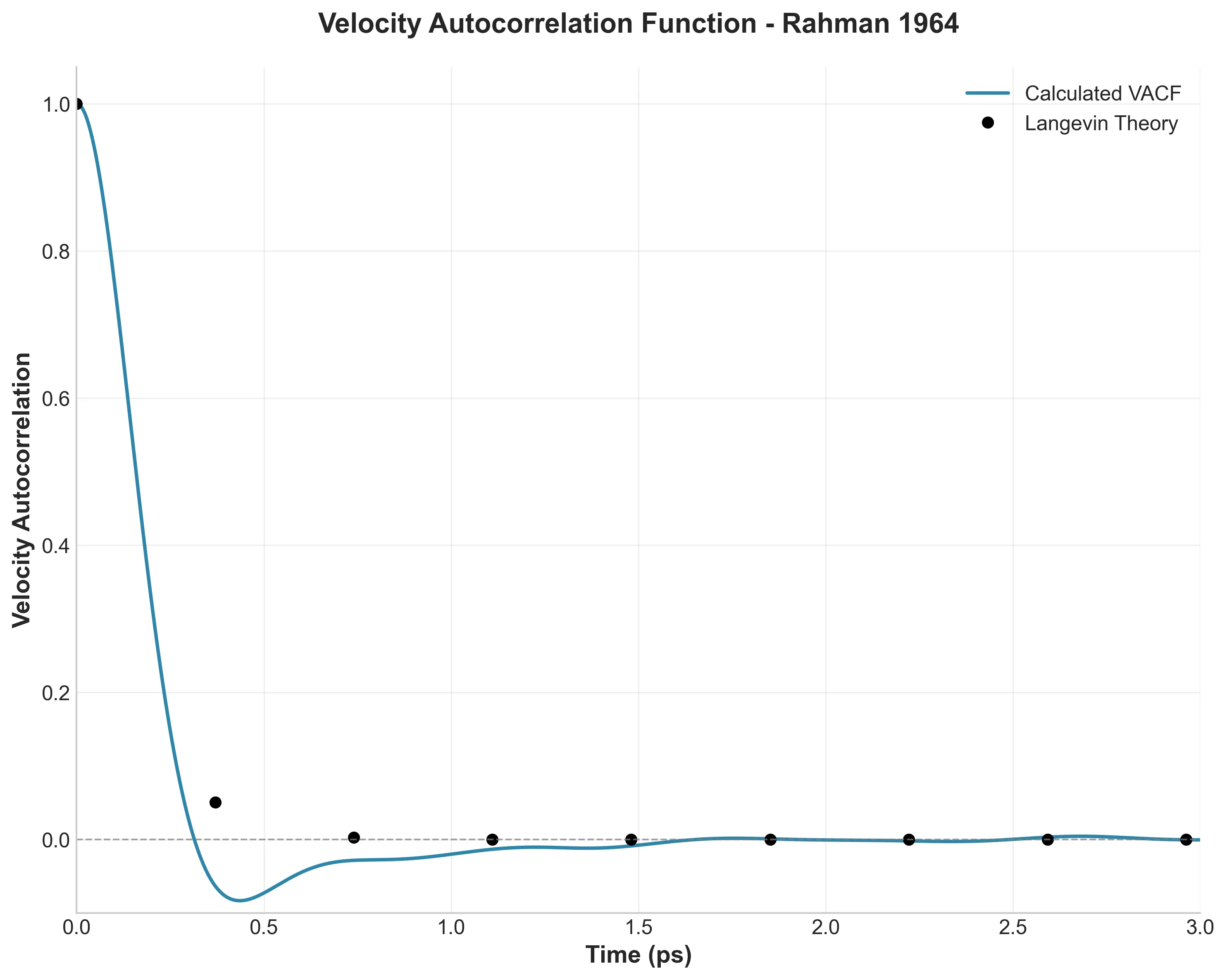
A PyTorch testbed for the Müller-Brown potential energy surface, built as ground truth for ML-in-molecular-dynamics work. It pairs analytical and autograd force kernels (the analytical path compiled with torch.compile) with a BAOAB Langevin sampler validated against the canonical distribution and a two-tier test suite.