GEOM: Energy-Annotated Molecular Conformations Dataset
GEOM contains 450k+ molecules with 37M+ conformations, featuring energy annotations from semi-empirical (GFN2-xTB) and DFT methods for property prediction and molecular generation research.
GEOM contains 450k+ molecules with 37M+ conformations, featuring energy annotations from semi-empirical (GFN2-xTB) and DFT methods for property prediction and molecular generation research.

Explore two fundamental approaches to generating exponentially distributed random numbers: the modern inverse transform method using logarithms and von Neumann’s ingenious 1951 comparison-based algorithm that avoids transcendental functions entirely.
GDB-11 contains 26.4 million systematically generated small organic molecules with up to 11 atoms, establishing the methodology for exploring drug-like chemical space computationally.
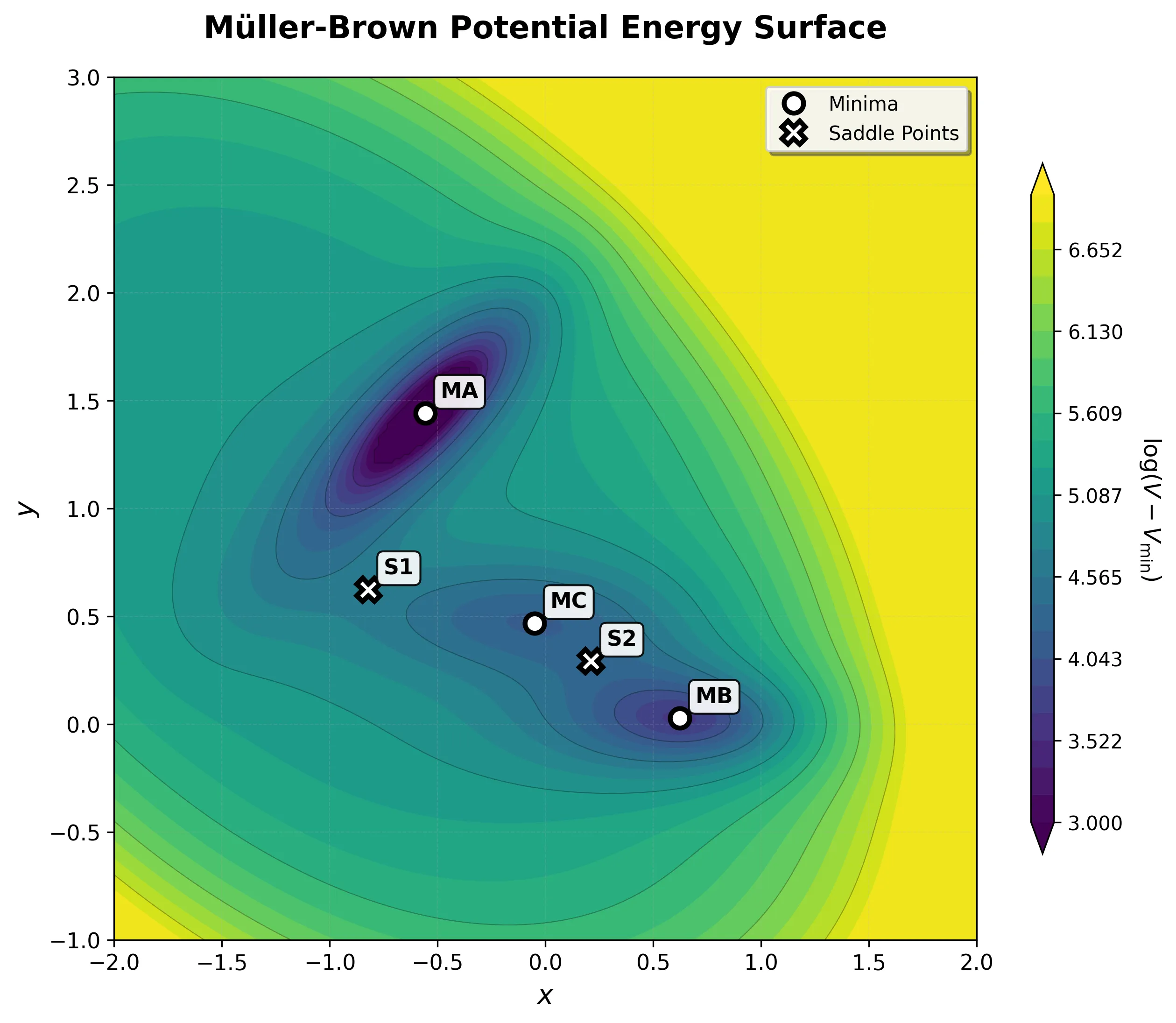
Step-by-step implementation of the classic Müller-Brown potential in PyTorch, with performance comparisons between analytical and automatic differentiation approaches for molecular dynamics and machine learning applications.
A high-performance, GPU-accelerated PyTorch testbed for ML-MD algorithms featuring JIT-compiled analytical Jacobian force kernels achieving 3-10x speedup over autograd, robust Langevin dynamics with Velocity-Verlet integration, and modular architecture designed as ground-truth validation for novel machine learning approaches in molecular dynamics.
ICLR 2025 paper introducing DenoiseVAE, which learns adaptive, atom-specific noise distributions through a VAE framework to improve denoising-based pre-training for molecular force field prediction, outperforming fixed Gaussian noise approaches on quantum chemistry benchmarks.
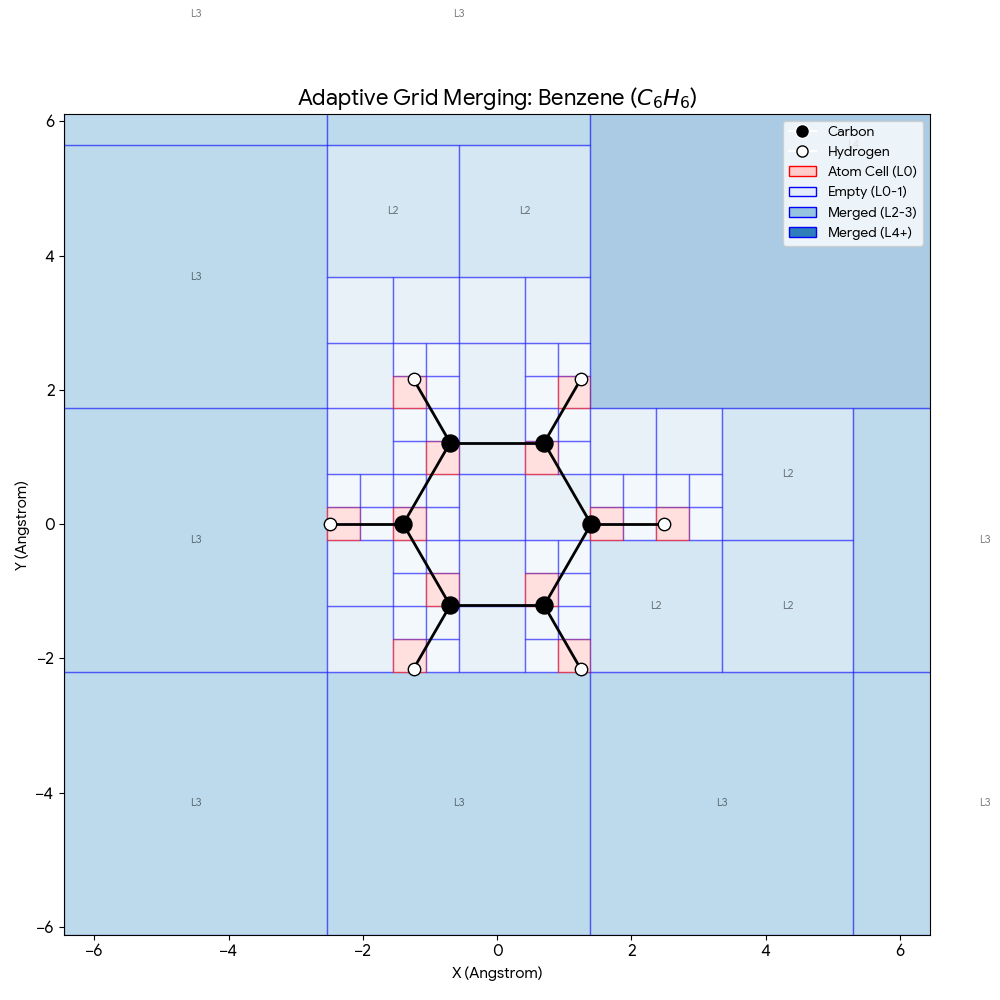
ICML 2025 paper introducing SpaceFormer, a Transformer architecture that challenges the atom-centric paradigm by modeling the continuous 3D space surrounding molecules using adaptive multi-resolution grids, ranking first in 10 of 15 molecular property prediction tasks.
ICML 2025 analysis rigorously quantifying when non-conservative force models (which predict forces directly) fail in molecular dynamics, demonstrating simulation instabilities and proposing hybrid architectures that capture speed benefits without sacrificing physical correctness.
ICML 2025 methodological paper introducing SPHNet, which uses adaptive network sparsification to overcome the computational bottleneck of tensor products in SE(3)-equivariant networks, achieving up to 7x speedup and 75% memory reduction on DFT Hamiltonian prediction tasks.
ICML 2025 paper proposing energy conservation metrics as critical diagnostics for machine learning interatomic potentials and introducing eSEN, a novel architecture designed to bridge the gap between test-set accuracy and real simulation performance on materials property prediction.
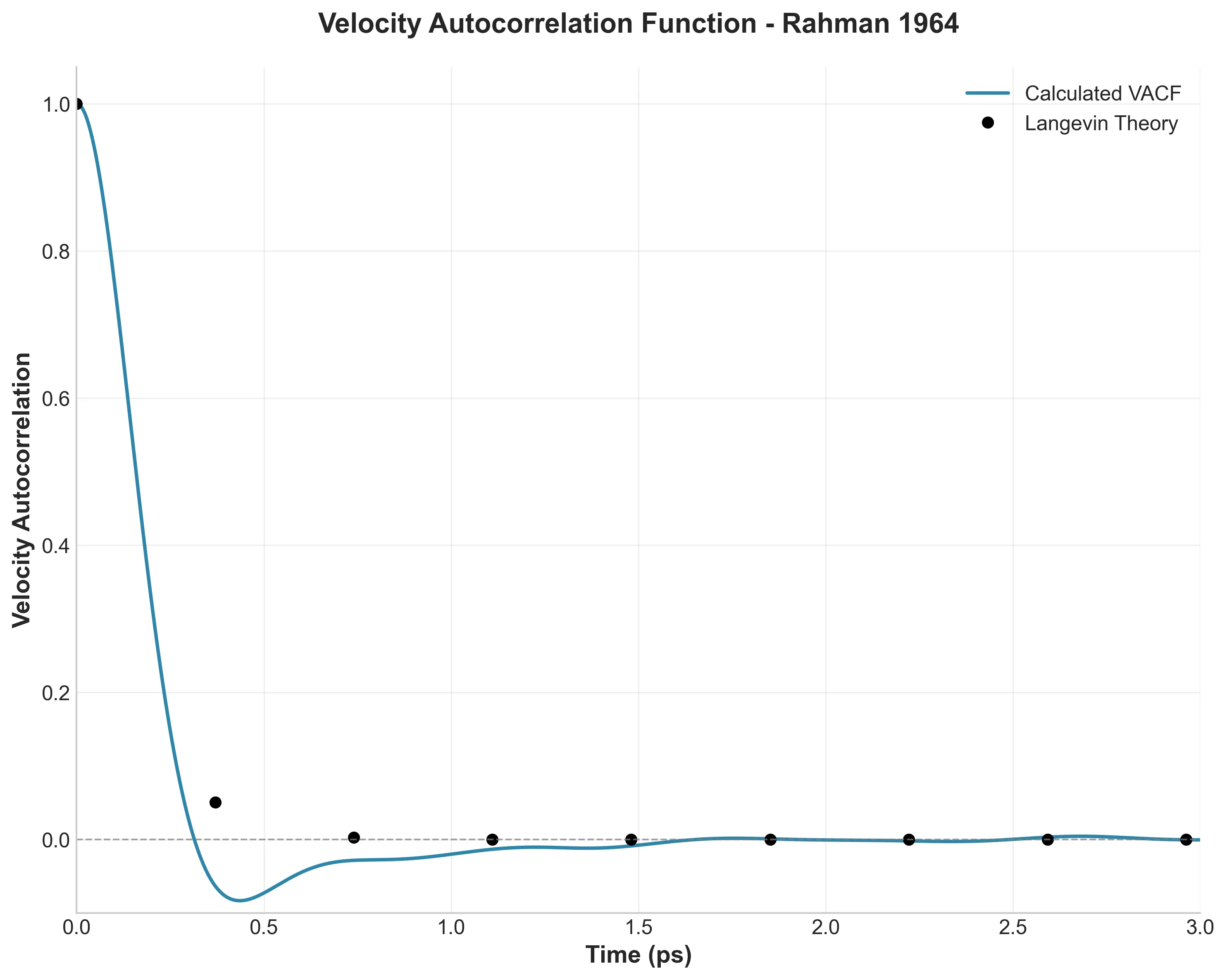
A digital restoration of Rahman’s seminal 1964 molecular dynamics paper using LAMMPS and a production-grade Python analysis pipeline featuring intelligent decorator-based caching, fully vectorized NumPy computations for O(N^2) operations, and modern tooling (uv, type hints, Makefile automation) transforming academic scripts into reproducible research toolkit.
I replicated Rahman’s landmark 1964 liquid argon molecular dynamics simulation using modern tools, building a production-grade Python analysis pipeline with intelligent caching, vectorization, and type safety to bridge vintage science with contemporary software engineering.