What kind of paper is this?
This is a discovery paper that identifies empirical neural scaling laws in two distinct domains of chemical deep learning: large language models (LLMs) for generative chemistry and graph neural networks (GNNs) for machine-learned interatomic potentials. The paper also introduces training performance estimation (TPE) as a practical tool for accelerating hyperparameter optimization in these domains.
Why scaling laws matter for chemistry
Neural scaling laws, first characterized for NLP models by Kaplan et al. (2020), describe how model loss decreases as a power law with increasing model size, dataset size, or compute:
$$ L(R) = \alpha R^{-\beta} $$
where $\alpha$ is a coefficient, $\beta$ is the scaling exponent, and $R$ is the resource being scaled (parameters, data, or compute). These relationships have guided resource allocation decisions in NLP and computer vision, but their applicability to scientific deep learning was unknown.
Chemical deep learning differs from standard NLP and vision tasks in several key ways. Physics-based priors (like symmetry constraints) may reduce the need for massive scale. The heterogeneity of chemical space and molecular tasks makes general pre-training more challenging. There are no established default architectures, datasets, or training recipes at large scale for chemistry.
This paper asks: do the same scaling behaviors hold for chemical models, and how do physical priors affect them?
Training performance estimation for efficient scaling
Before running expensive scaling experiments, the authors needed a way to efficiently select hyperparameters. They introduced TPE, a generalization of training speed estimation (TSE) to new domains. TSE computes the cumulative training loss over the first $T$ epochs:
$$ \text{TSE} = \sum_{t=1}^{T} \left( \frac{1}{B} \sum_{i=1}^{B} \mathcal{L}\left(f_{\theta(t,i)}(\mathbf{X}_i), \mathbf{y}_i\right) \right) $$
where $B$ is the number of training steps per epoch, $\mathcal{L}$ is the loss function, and $f_{\theta(t,i)}$ is the network at epoch $t$ and mini-batch $i$. A linear regression then predicts converged loss from early-training TSE:
$$ L = m \times \text{TSE} + b $$
Using only 20% of the total training budget, TPE achieves $R^2 = 0.98$ and Spearman’s $\rho = 1.0$ for ChemGPT on the MOSES dataset. For GNNs, it achieves $R^2 \geq 0.86$ and $\rho \geq 0.92$ across SchNet, PaiNN, and SpookyNet. This enables discarding suboptimal configurations early, saving up to 90% of compute.
ChemGPT: scaling chemical language models
ChemGPT is a GPT-3-style autoregressive transformer for molecular generation. It uses GPT-Neo as its backbone with a SELFIES tokenizer, factorizing the probability of a molecular sequence as:
$$ p(x) = \prod_{i=1}^{n} p\left(s_i \mid s_1, \dots, s_{i-1}\right) $$
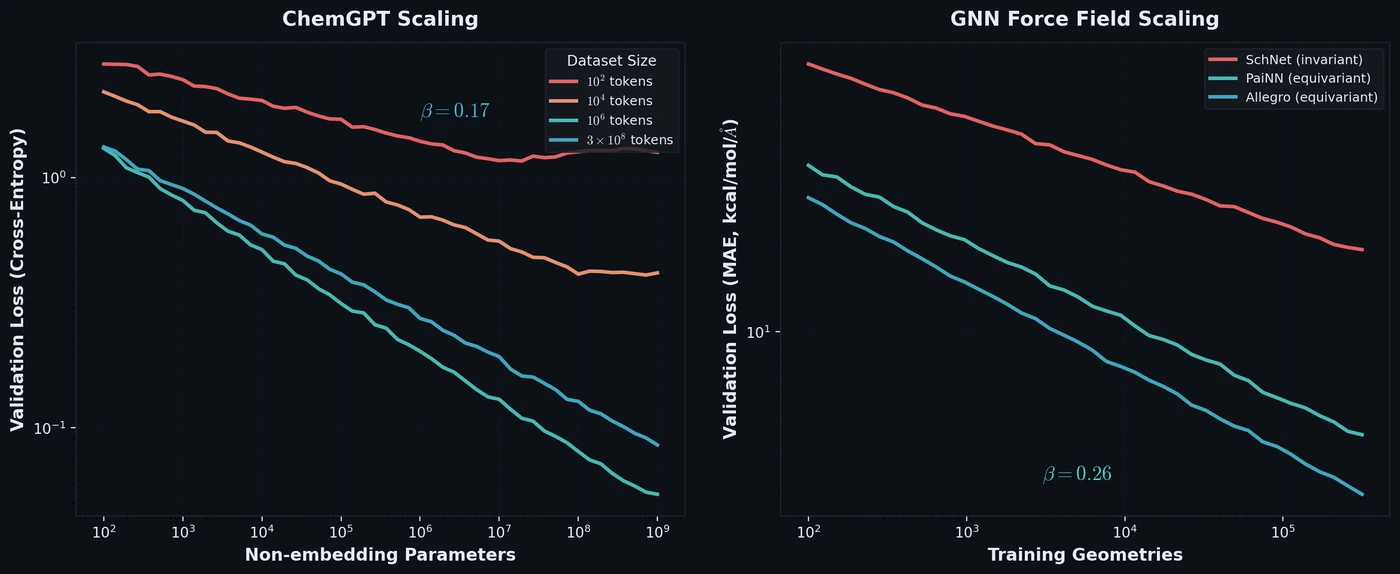
The authors trained ChemGPT models ranging from ~78K to over 1 billion non-embedding parameters on subsets of PubChem10M (up to ~10 million molecules, or ~300 million tokens). Key findings from the scaling experiments:
- Pre-training loss monotonically improves with increasing dataset size up to nearly 10 million molecules, with no saturation observed.
- For a fixed data budget, increasing model size provides monotonic improvements until models reach ~1 billion parameters.
- The scaling exponent $\beta = 0.17 \pm 0.01$ for the largest dataset (after excluding the three largest models from the power-law fit), and $\beta = 0.30 \pm 0.01$ for the next largest dataset.
- Resolution-limited regimes appear where the power-law behavior breaks down, indicating either insufficient data for a given model size or vice versa. These regimes shift depending on the data budget.
An interesting observation: for small datasets, large models ($10^7$ parameters and above) still provide notable loss improvements, suggesting that scaling up model size helps even when data is limited.
Neural force field scaling with GNNs
For tasks requiring three-dimensional molecular geometry, the authors studied GNN-based neural force fields (NFFs). These models predict energies $\hat{E} = f_\theta(X)$ and derive forces by differentiation:
$$ \hat{F}_{ij} = -\frac{\partial \hat{E}}{\partial r_{ij}} $$
Training uses an L1 loss over energies and forces:
$$ \mathcal{L} = \frac{1}{N} \sum_{i=1}^{N} \left[ \alpha_E | E_i - \hat{E}_i | + \alpha_F | \mathbf{F}_i - \hat{\mathbf{F}}_i | \right] $$
Four NFF architectures were studied, spanning a range of physical priors:
| Model | Type | Key Characteristic |
|---|---|---|
| SchNet | E(3) invariant | Continuous filter convolutions |
| PaiNN | E(3) equivariant | Equivariant message passing |
| Allegro | E(3) equivariant | Local, learned many-body functions |
| SpookyNet | E(3) equivariant | Non-local interactions, empirical corrections |
Model capacity is parameterized as $c = d \times w$ (depth times width). Models were trained on subsets of the ANI-1x dataset (up to 100,000 geometries, corresponding to ~4.5 million force labels).
Key GNN scaling findings:
- PaiNN shows monotonic loss improvement with increasing dataset size and strong correlation between converged loss and model capacity (Spearman’s $\rho \geq 0.88$).
- Equivariant GNNs (PaiNN, Allegro) show better scaling efficiency than invariant GNNs (SchNet), with larger $\beta$ values.
- The scaling exponent for equivariant GNNs is $\beta = 0.26$, indicating that physics-based equivariance priors provide greater sample efficiency that persists to much larger and more chemically diverse datasets than previously studied.
- A transition at $10^4$ datapoints shows nearly perfect rank correlation between model capacity and converged loss ($\rho \geq 0.93$), suggesting this may be a threshold where models move from memorization to generalization.
Results and practical implications
The scaling results provide actionable guidance for resource allocation:
- For chemical LLMs with large data budgets, the greatest loss improvements come from scaling up small models (around $10^5$ parameters).
- For small data budgets, rapid improvements come from scaling medium-sized models ($10^7$ parameters).
- For NFFs, low-capacity models show diminishing returns with more data, while high-capacity models show rapid improvements with increasing dataset size.
- Neither model type has saturated with respect to model size, dataset size, or compute, suggesting substantial room for improvement with further scaling.
The 300-million-parameter ChemGPT trained on 300 million tokens and the PaiNN model with capacity ~1,000 trained on $10^5$ frames achieved the minimum losses in their respective scaling plots, providing concrete targets for practitioners.
Reproducibility Details
Data:
- PubChem10M (10M SMILES strings, via DeepChem)
- MOSES (2M molecules, for TPE validation)
- ANI-1x (5M DFT calculations, via Figshare)
- Revised MD-17 (10 small organic molecules, 10,000 frames for TPE)
Models:
- ChemGPT: GPT-Neo backbone, 24 layers, widths from 16 to 2,048, sizes from ~78K to ~1.2B non-embedding parameters
- SchNet, PaiNN, Allegro, SpookyNet: widths of 16, 64, 256; depths of 2, 3, 4; 5 Angstrom cutoff
Training:
- ChemGPT: AdamW optimizer, learning rate $2 \times 10^{-5}$, batch size 8 per GPU, 10 epochs, cross-entropy loss
- GNNs: Adam optimizer, learning rate scheduler (halved after 30 epochs without improvement), early stopping after 50 stagnant epochs, max 1,000 epochs, L1 loss (force-only training)
Hardware:
- NVIDIA Volta V100 GPUs (32 GB), 2 GPUs per node
- PyTorch with distributed data parallel (DDP), PyTorch Lightning, LitMatter
Code: LitMatter repository
Paper Information
Citation: Frey, N.C., Soklaski, R., Axelrod, S. et al. Neural scaling of deep chemical models. Nat Mach Intell 5, 1297-1305 (2023).
@article{frey2023neural,
title={Neural scaling of deep chemical models},
author={Frey, Nathan C. and Soklaski, Ryan and Axelrod, Simon and Samsi, Siddharth and G{\'o}mez-Bombarelli, Rafael and Coley, Connor W. and Gadepally, Vijay},
journal={Nature Machine Intelligence},
volume={5},
pages={1297--1305},
year={2023},
publisher={Nature Publishing Group},
doi={10.1038/s42256-023-00740-3}
}