What kind of paper is this?
This is a Methodological ($\Psi_{\text{Method}}$) paper. It introduces a novel architecture, InvMSAFold, which hybridizes deep learning encoders with statistical physics-based decoders (Potts models). The rhetorical structure focuses on architectural innovation (low-rank parameter generation), ablation of speed/diversity against baselines (ESM-IF1), and algorithmic efficiency.
What is the motivation?
Standard inverse folding models (like ESM-IF1 or ProteinMPNN) solve a “one-to-one” mapping: given a structure, predict the single native sequence. However, in nature, folding is “many-to-one”: many homologous sequences fold into the same structure.
The authors identify two key gaps:
- Lack of Diversity: Standard autoregressive models maximize probability for the ground truth sequence, often failing to capture the broad evolutionary landscape of viable homologs.
- Slow Inference: Autoregressive sampling requires a full neural network pass for every amino acid, making high-throughput screening (e.g., millions of candidates) computationally prohibitive.
What is the novelty here?
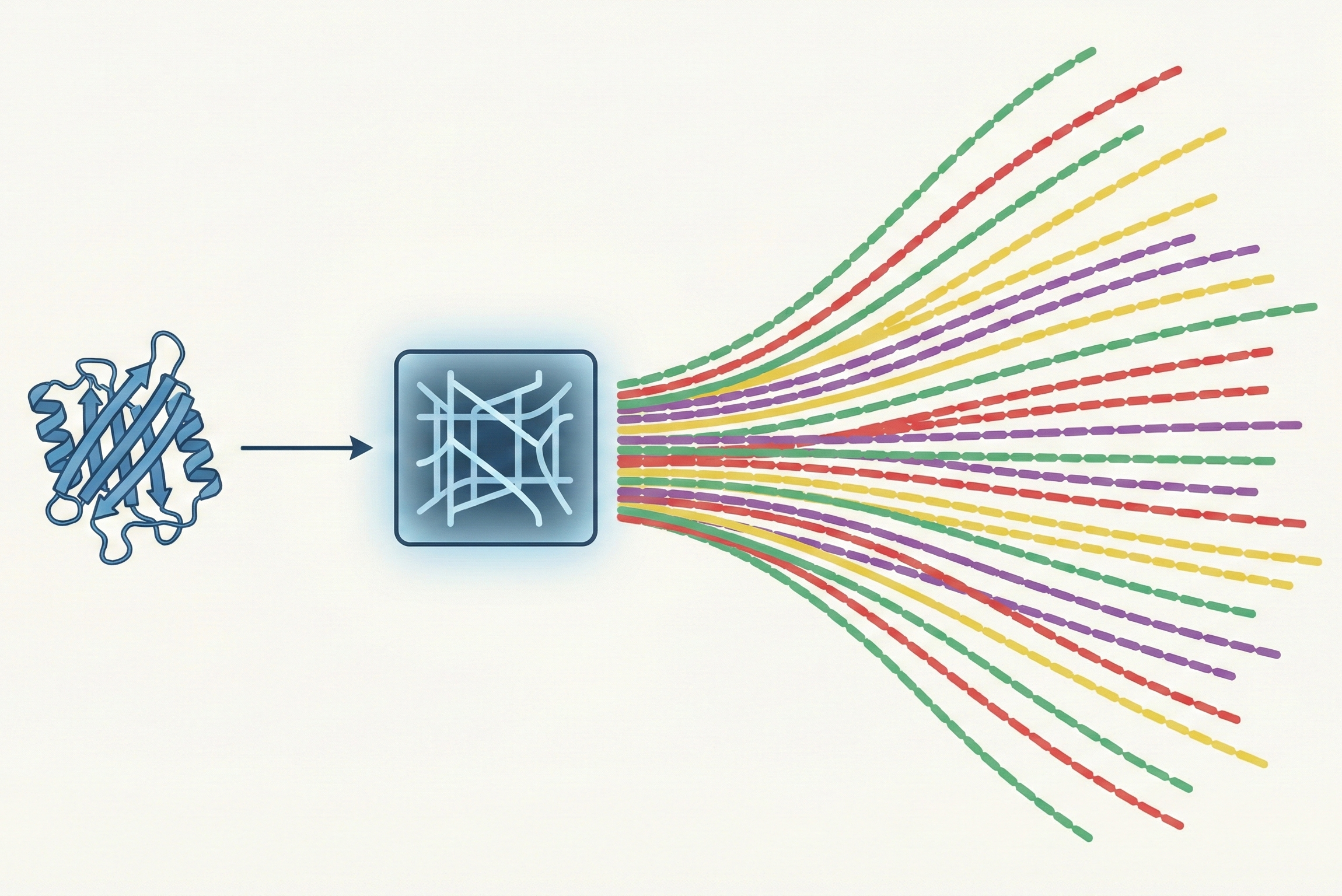
The core novelty is shifting the learning objective from predicting sequences to predicting probability distributions.
InvMSAFold outputs the parameters (couplings $\mathbf{J}$ and fields $\mathbf{h}$) of a Potts Model (a pairwise Markov Random Field).
- Low-Rank Decomposition: To handle the massive parameter space of pairwise couplings ($L \times L \times q \times q$), the model predicts a low-rank approximation $\mathbf{V}$ ($L \times K \times q$), reducing complexity from $\mathcal{O}(L^2)$ to $\mathcal{O}(L)$.
- One-Shot Generation: The deep network runs only once to generate the Potts parameters. Sampling sequences from this Potts model is then performed on CPU via MCMC (for the PW variant) or direct autoregressive sampling (for the AR variant), which is orders of magnitude faster than running a Transformer decoder for every step.
What experiments were performed?
The authors validated the model on three CATH-based test sets (Inter-cluster, Intra-cluster, MSA) to test generalization at varying levels of homology.
- Speed Benchmarking: Compared wall-clock sampling time vs. ESM-IF1 on CPU/GPU.
- Covariance Reconstruction: Checked if generated sequences recover the evolutionary correlations found in natural MSAs (Pearson correlation of covariance matrices).
- Structural Fidelity: Generated sequences with high Hamming distance from native, folded them with AlphaFold 2 (no templates), and measured RMSD to the target structure.
- Property Profiling: Analyzed the distribution of predicted solubility (Protein-Sol) and thermostability (Thermoprot) to show that sequence diversity translates into a wider range of biochemical properties.
What outcomes/conclusions?
- Massive Speedup: InvMSAFold is orders of magnitude faster than ESM-IF1 (CPU vs. GPU; the comparison is not hardware-matched). Because the “heavy lifting” (generating Potts parameters) happens once, sampling millions of sequences becomes trivial on CPUs.
- Better Diversity: The model captures evolutionary covariances significantly better than ESM-IF1 and ProteinMPNN (which shares similar covariance recovery to ESM-IF1). A PCA-based KL-divergence analysis (lower is better; 0 means a perfect match to the natural MSA distribution) shows InvMSAFold-AR scores of $0.49$ (Inter-cluster) and $0.67$ (Intra-cluster), compared to $15.8$ and $11.9$ for ESM-IF1, demonstrating that the generated sequences occupy a distribution much closer to natural MSAs.
- Robust Folding: Sequences generated far from the native sequence (high Hamming distance) still fold into the correct structure (low RMSD), whereas ESM-IF1 struggles to produce diverse valid sequences.
- Property Expansion: The method generates a wider spread of predicted biochemical properties (solubility/thermostability), which could be useful for virtual screening in protein design.
Reproducibility Details
Data
Source: CATH database (40% non-redundant dataset).
Splits:
- Training: ~22k domains.
- Inter-cluster Test: 10% of sequence clusters held out (unseen clusters, many with superfamilies absent from training).
- Intra-cluster Test: Unseen domains from seen clusters.
- Augmentation: MSAs generated using MMseqs2 against the Uniprot50 database. Training uses random subsamples of these MSAs ($|M_X| = 64$ for PW, $|M_X| = 32$ for AR) to teach the model evolutionary variance.
Algorithms
Architecture:
- Encoder: Pre-trained ESM-IF1 encoder (GVP-GNN architecture). The encoder is used to pre-compute structure embeddings, with independent Gaussian noise (std = 5% of the embedding std) added during training.
- Decoder: 6-layer Transformer (8 heads) that outputs a latent tensor.
- Projection: Linear layers project latent tensor to fields $\mathbf{h}$ ($L \times q$) and low-rank tensor $\mathbf{V}$ ($L \times K \times q$).
Coupling Construction: The full coupling tensor $\mathcal{J}$ is approximated via: $$\mathcal{J}_{i,a,j,b} = \frac{1}{\sqrt{K}} \sum_{k=1}^{K} \mathcal{V}_{i,k,a} \mathcal{V}_{j,k,b}$$ Rank $K=48$ was used.
Loss Functions: Two variants were trained:
- InvMSAFold-PW: Trained via Pseudo-Likelihood (PL). Computation is optimized to $\mathcal{O}(L)$ time using the low-rank property.
- InvMSAFold-AR: Trained via Autoregressive Likelihood. Couplings are masked ($J_{ij} = 0$ if $i < j$) to allow exact likelihood computation and direct sampling without MCMC.
Models
- InvMSAFold-PW: Requires MCMC sampling (Metropolis-Hastings) at inference.
- InvMSAFold-AR: Allows direct, fast autoregressive sampling.
- Hyperparameters: AdamW optimizer, lr=$10^{-4}$ (PW) / $3.4 \times 10^{-4}$ (AR), 94 epochs. L2 regularization: $\lambda_h = \lambda_J = 10^{-4}$ (PW); $\lambda_J = 3.2 \times 10^{-6}$, $\lambda_h = 5.0 \times 10^{-5}$ (AR).
Evaluation
Metrics:
- RMSD: Structure fidelity (AlphaFold2 prediction vs. native structure).
- Covariance Pearson Correlation: Measures recovery of evolutionary pairwise statistics.
- KL Divergence: Between PCA-projected densities of natural and synthetic sequences (Gaussian KDE, kernel size 1.0).
- Sampling Speed: Wall-clock time vs. sequence length/batch size.
Hardware
- Training: Not specified in the paper. The GitHub repository reports testing on an NVIDIA RTX 3090, with training taking 10-24 hours depending on model variant.
- Inference:
- ESM-IF1: NVIDIA GeForce RTX 4060 Laptop (8GB).
- InvMSAFold: Single core of Intel i9-13905H CPU.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| Potts_Inverse_Folding | Code | MIT | Training and inference code (PyTorch) |
Paper Information
Citation: Silva, L. A., Meynard-Piganeau, B., Lucibello, C., & Feinauer, C. (2025). Fast Uncovering of Protein Sequence Diversity from Structure. International Conference on Learning Representations (ICLR). https://arxiv.org/abs/2406.11975
Publication: ICLR 2025 (Spotlight)
@inproceedings{silvaFastUncoveringProtein2025,
title = {Fast {{Uncovering}} of {{Protein Sequence Diversity}} from {{Structure}}},
booktitle = {The {{Thirteenth International Conference}} on {{Learning Representations}}},
author = {Silva, Luca Alessandro and {Meynard-Piganeau}, Barthelemy and Lucibello, Carlo and Feinauer, Christoph},
year = {2025},
url = {https://openreview.net/forum?id=1iuaxjssVp}
}
Additional Resources: