What kind of paper is this?
This is primarily a Methodological Paper ($\Psi_{\text{Method}}$) with a strong Resource ($\Psi_{\text{Resource}}$) component.
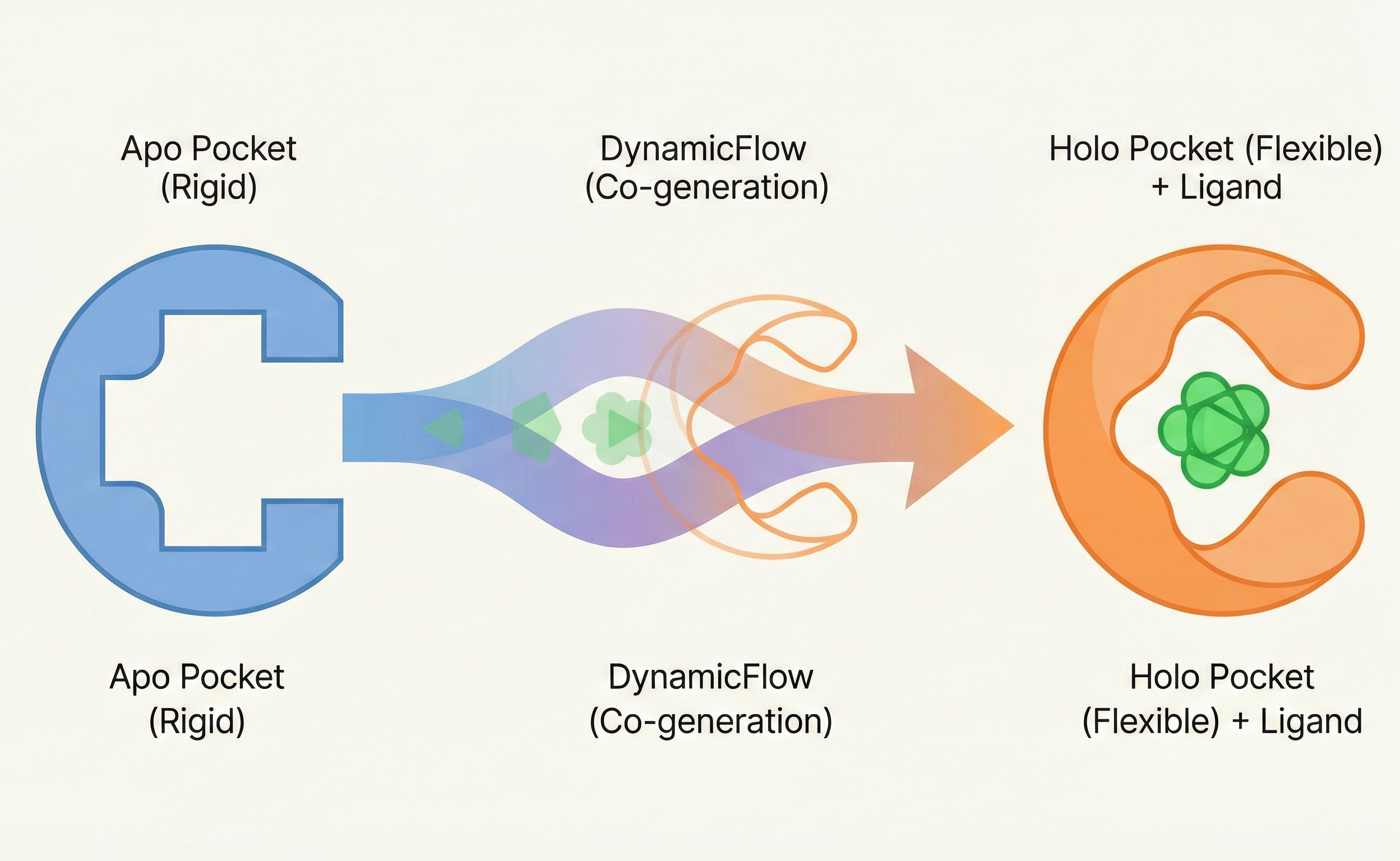
- Method: It proposes DynamicFlow, a novel multiscale architecture combining atom-level SE(3)-equivariant GNNs (SE(3) is the special Euclidean group in 3D: the set of all 3D rotations and translations, and equivariance means predictions transform consistently under those symmetries) and residue-level Transformers within a flow matching framework to model the joint distribution of ligand generation and protein conformational change.
- Resource: It curates a significant dataset derived from MISATO, pairing AlphaFold2-predicted apo structures with multiple MD-simulated holo states, specifically filtered for flow matching tasks.
What is the motivation?
Traditional Structure-Based Drug Design (SBDD) methods typically assume the protein target is rigid, which limits their applicability because proteins are dynamic and undergo conformational changes (induced fit) upon ligand binding.
- Biological Reality: Proteins exist as ensembles of states; binding often involves transitions from “apo” (unbound) to “holo” (bound) conformational changes, sometimes revealing cryptic pockets.
- Computational Bottleneck: Molecular Dynamics (MD) simulates these changes but incurs high computational costs due to energy barriers.
- Gap: Existing generative models for SBDD mostly condition on a fixed pocket structure, ignoring the co-adaptation of the protein and ligand.
What is the novelty here?
The core novelty is the simultaneous modeling of ligand generation and protein conformational dynamics using a unified flow matching framework.
- DynamicFlow Architecture: A multiscale model that treats the protein as both full-atom (for interaction) and residue-level frames (for large-scale dynamics), utilizing separate flow matching objectives for backbone frames, side-chain torsions, and ligand atoms.
- Stochastic Flow (SDE): Introduction of a stochastic variant (DynamicFlow-SDE) that improves robustness and diversity compared to the deterministic ODE flow.
- Coupled Generation: The model learns to transport the apo pocket distribution to the holo pocket distribution while simultaneously denoising the ligand, advancing beyond rigid pocket docking methods.
What experiments were performed?
The authors validated the method on a curated dataset of 5,692 protein-ligand complexes.
- Baselines: Compared against rigid-pocket SBDD methods: Pocket2Mol, TargetDiff, and IPDiff (adapted as TargetDiff* and IPDiff* for fair comparison of atom numbers). Also compared against conformation sampling baselines (Str2Str).
- Metrics:
- Ligand Quality: Vina Score (binding affinity), QED (drug-likeness), SA (synthesizability), Lipinski’s rule of 5.
- Pocket Quality: RMSD between generated and ground-truth holo pockets, Cover Ratio (percentage of holo states successfully retrieved), and Pocket Volume distributions.
- Interaction: Protein-Ligand Interaction Profiler (PLIP) to measure specific non-covalent interactions.
- Ablations: Tested the impact of the interaction loss, residue-level Transformer, and SDE vs. ODE formulations.
What outcomes/conclusions?
- Improved Affinity: DynamicFlow-SDE achieved the best (lowest) Vina scores ($-7.65$) compared to baselines like TargetDiff ($-5.09$) and Pocket2Mol ($-5.50$). Note that Vina scores are a computational proxy and do not directly predict experimental binding affinity. Moreover, Vina score optimization is gameable: molecules can achieve strong computed binding energies while remaining synthetically inaccessible. QED and SA scores, which assess drug-likeness and synthesizability respectively, were reported but were not primary optimization targets in the paper, which limits the strength of this affinity claim.
- Realistic Dynamics: The model successfully generated holo-like pocket conformations with volume distributions and interaction profiles closer to ground-truth MD simulations than the initial apo structures.
- Enhancing Rigid Methods: Holo pockets generated by DynamicFlow served as better inputs for rigid-SBDD baselines (e.g., TargetDiff improved from $-5.09$ to $-9.00$ and IPDiff improved from $-7.55$ to $-11.04$ when using “Our Pocket”), suggesting the method can act as a “pocket refiner”.
- ODE vs. SDE Trade-off: The deterministic ODE variant achieves better pocket RMSD, while the stochastic SDE variant achieves better Cover Ratio (diversity of holo states captured) and binding affinity. Neither dominates uniformly.
- Conformation Sampling Baseline: Str2Str, a dedicated conformation sampling baseline, performed worse than simply perturbing the apo structure with noise. One interpretation is that this highlights the difficulty of the apo-to-holo prediction task; another is that Str2Str was not designed specifically for apo-to-holo prediction, making it a limited test of its capabilities.
Reproducibility Details
Data
The dataset is derived from MISATO, which contains MD trajectories for PDBbind complexes.
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training/Test | Curated MISATO | 5,692 complexes | Filtered for valid MD (RMSD $< 3\text{\AA}$), clustered to remove redundancy. Contains 46,235 holo-ligand conformations total. |
| Apo Structures | AlphaFold2 | N/A | Apo structures were obtained by mapping PDB IDs to UniProt and retrieving AlphaFold2 predictions, then aligning to MISATO structures. |
| Splits | Standard | 50 test complexes | 50 complexes with no overlap with the training set selected for testing. Note: 50 is a small held-out set; results should be interpreted cautiously. |
Preprocessing:
- Clustering: Holo-ligand conformations clustered with RMSD threshold $1.0\text{\AA}$; top 10 clusters kept per complex.
- Pocket Definition: Residues within $7\text{\AA}$ of the ligand.
- Alignment: AlphaFold predicted structures (apo) aligned to MISATO holo structures using sequence alignment (Smith-Waterman) to identify pocket residues.
Algorithms
Flow Matching Framework:
- Continuous Variables (Pocket translation/rotation/torsions, Ligand positions): Modeled using Conditional Flow Matching (CFM).
- Prior: Apo state for pocket; Normal distribution for ligand positions.
- Target: Holo state from MD; Ground truth ligand.
- Interpolant: Linear interpolation for Euclidean variables; Geodesic for rotations ($SO(3)$, the rotation-only subgroup of SE(3) containing all 3D rotations but not translations); Wrapped linear interpolation for torsions (Torus).
- Discrete Variables (Ligand atom/bond types): Modeled using Discrete Flow Matching based on Continuous-Time Markov Chains (CTMC).
- Rate Matrix: Interpolates between mask token and data distribution.
- Loss Function: Weighted sum of 7 losses:
- Translation CFM (Eq 5)
- Rotation CFM (Eq 7)
- Torsion CFM (Eq 11)
- Ligand Position CFM
- Ligand Atom Type CTMC (Eq 14)
- Ligand Bond Type CTMC
- Interaction Loss (Eq 18): Explicitly penalizes deviations in pairwise distances between protein and ligand atoms for pairs $\leq 3.5\text{\AA}$.
Models
Architecture: DynamicFlow is a multiscale model with 15.9M parameters.
- Atom-Level SE(3)-Equivariant GNN:
- Input: Complex graph (k-NN) and Ligand graph (fully connected).
- Layers: 6 EGNN blocks modified to maintain node and edge hidden states.
- Function: Updates ligand positions and predicts ligand atom/bond types.
- Residue-Level Transformer:
- Input: Aggregated atom features from the GNN + Residue frames/torsions.
- Layers: 4 Transformer blocks with Invariant Point Attention (IPA).
- Function: Updates protein residue frames (translation/rotation) and predicts side-chain torsions.
Evaluation
Metrics:
- Vina Score:
vina_minimizemode used for binding affinity. - RMSD: Minimum RMSD between generated pocket and ground-truth holo conformations.
- Cover Ratio: % of ground-truth holo conformations covered by at least one generated sample (threshold $1.42\text{\AA}$).
- POVME 3: For pocket volume calculation.
Hardware
- Inference Benchmark: 1x Tesla V100-SXM2-32GB.
- Speed: Generates 10 ligands in ~35-36 seconds (100 NFE), significantly faster than diffusion baselines like Pocket2Mol (980s) or TargetDiff (156s).
Paper Information
Citation: Zhou, X., Xiao, Y., Lin, H., He, X., Guan, J., Wang, Y., Liu, Q., Zhou, F., Wang, L., & Ma, J. (2025). Integrating Protein Dynamics into Structure-Based Drug Design via Full-Atom Stochastic Flows. International Conference on Learning Representations (ICLR). https://arxiv.org/abs/2503.03989
Publication: ICLR 2025
@inproceedings{zhouIntegratingProteinDynamics2025,
title = {Integrating Protein Dynamics into Structure-Based Drug Design via Full-Atom Stochastic Flows},
author = {Zhou, Xiangxin and Xiao, Yi and Lin, Haowei and He, Xinheng and Guan, Jiaqi and Wang, Yang and Liu, Qiang and Zhou, Feng and Wang, Liang and Ma, Jianzhu},
booktitle = {International Conference on Learning Representations},
year = {2025},
url = {https://arxiv.org/abs/2503.03989}
}
Additional Resources:
- arXiv Page
- Code: no public repository available at time of writing