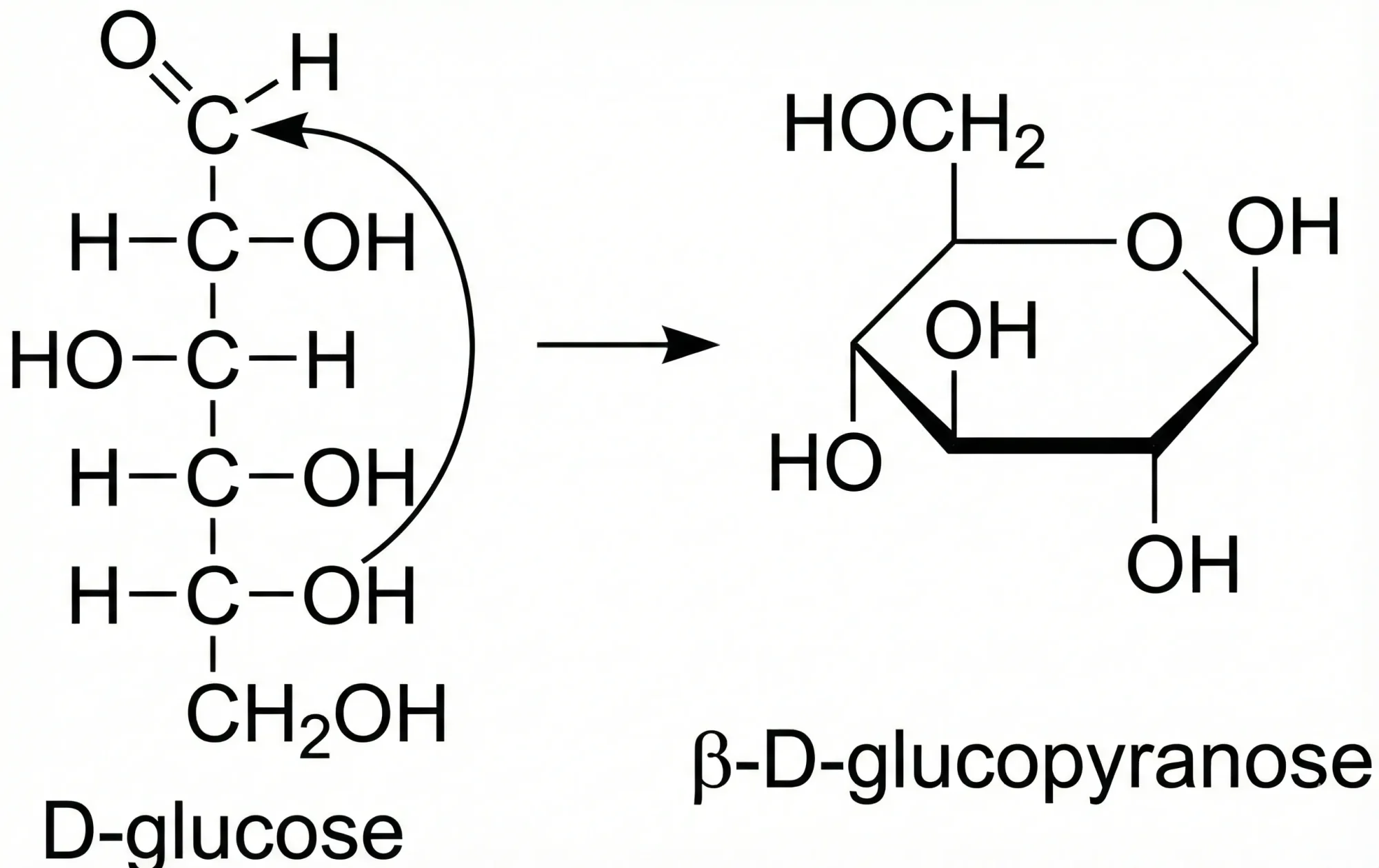
InChI and Tautomerism: Toward Comprehensive Treatment
Dhaked et al.'s comprehensive analysis of tautomerism in chemoinformatics, introducing 86 new tautomeric rules and their …
Dhaked et al.'s comprehensive analysis of tautomerism in chemoinformatics, introducing 86 new tautomeric rules and their …
Heller et al. (2013) explain how IUPAC's InChI became the global standard for representing chemical structures, its …
InChI v1.07 modernizes chemical identifiers for FAIR principles and adds robust support for inorganic compounds.

Mixfile and MInChI provide the first standardized, machine-readable formats for representing chemical mixtures.

NInChI (Nanomaterials InChI) extends chemical identifiers to represent complex, multi-component nanomaterials.
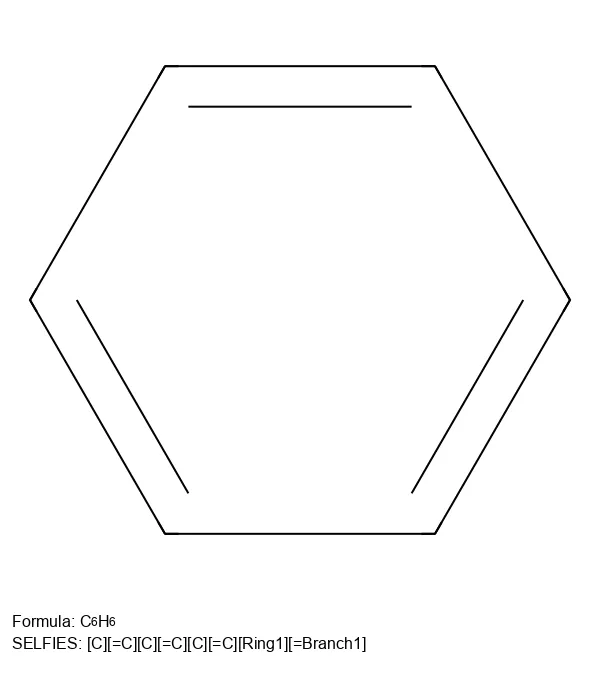
Major updates to the SELFIES library, improved performance, expanded chemistry support, and new customization features.
RInChI extends InChI to create unique, machine-readable identifiers for chemical reactions and database searching.
The 2020 paper introducing SELFIES, the 100% robust molecular representation that solves SMILES validity problems in ML …
Weininger's 1988 paper introducing SMILES notation, the string-based molecular representation that revolutionized …
SELFIES is a 100% robust molecular string representation for ML, implemented in the open-source selfies Python library.
SMILES (Simplified Molecular Input Line Entry System) represents chemical structures using compact ASCII strings.
Skinnider (2024) shows that generating invalid SMILES actually improves chemical language model performance through …